INTRODUCTION: The TCP
protein family is a plant-specific group of transcription factors known to
regulate key biological processes, including growth, development, and stress
responses. Despite their critical roles, the TCP gene family in Bergenia
purpurascens remains uncharacterized. This study aims to systematically
identify and analyze the BpTCP gene family in B. purpurascens using
transcriptome-based bioinformatics approaches, providing insights into their
potential functions in cold adaptation and secondary metabolism.
RATIONALE: B.
purpurascens exhibits remarkable resilience to abiotic
stresses, particularly cold, and contains abundant secondary metabolites. Given
the documented roles of TCP genes in stress responses and metabolic
regulation in other plants, we hypothesized that BpTCP genes may contribute
to these traits. A comprehensive analysis of this gene family could reveal
novel mechanisms underlying stress adaptation and metabolite synthesis,
supporting future genetic improvement or biotechnological applications.
RESULTS: Through transcriptome-based
bioinformatics analysis, we identified 16 BpTCP genes in B.
purpurascens, which were phylogenetically classified into two major groups,
with all members containing conserved TCP domains and closely related proteins
sharing similar motif patterns. Tissue-specific expression profiling revealed
distinct spatial expression patterns across different tissues, suggesting
functional diversification among family members. Notably, partial genes,
including BpTCP10, BpTCP1 and BpTCP12, exhibited
significant expression changes under cold stress, implying their potential
cold-responsive roles. Furthermore, expression levels of specific BpTCP genes correlated significantly with accumulation of various secondary
metabolites, particularly flavonoids and phenolics, suggesting their regulatory
involvement in metabolic pathways.
CONCLUSION: This study provides the first
genome-wide characterization of the BpTCP gene family in B.
purpurascens, demonstrating its potential roles in growth, cold stress
response, and secondary metabolism. The differential expression of BpTCP genes under stress and their correlation with metabolite levels lay a
foundation for future functional studies.
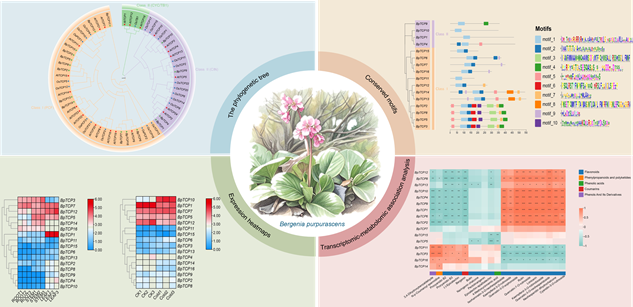
Expression pattern
and metabolic correlation analysis of TCP gene family in Bergenia
purpurascens. A total of 16 BpTCP genes were identified in Bergenia purpurascens and classified into two
major phylogenetic groups. All BpTCP genes contain conserved TCP
domains, and proteins from the same evolutionary branch share similar motif
compositions. Different BpTCP genes exhibit distinct tissue-specific
expression patterns and display distinctive responses to cold stress.
Furthermore, certain BpTCP genes demonstrate significant correlations
with the accumulation of diverse metabolites.


 Home
Home

