|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
重要林木樟科植物全基因组测序研究进展
植物学报
2024, 59 (2):
302-318.
DOI: 10.11983/CBB23035
近年来, 随着测序技术的革新、测序成本的降低和生物信息学软件的开发, 植物全基因组研究蓬勃发展。樟科(Lauraceae)隶属被子植物木兰类, 泛热带分布, 物种多样性高, 其中很多物种具有重要的经济和生态价值, 目前已发表包括8个物种的13个基因组。该文从樟科全基因组研究现状、基因组特征、起源和进化以及功能基因和基因家族4个方面进行综述, 着重介绍基于组学数据的木兰类及樟科的系统发生、樟科经历的多倍化事件以及与樟科花器官进化和代谢产物相关的基因鉴定。结合研究现状展望了樟科基因组研究的发展方向, 建议通过增加测序基因组分支的代表性并关注具有特殊价值的物种, 及研究物种特异性功能基因以加深对该家族基因功能和进化的理解。
表1
已测序樟科基因组信息
正文中引用本图/表的段落
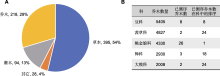
随着测序技术的进步、测序成本的降低以及全球经济的发展, 全基因组研究有加速的趋势。自第1个植物基因组序列被报道(The Arabidopsis Genome Initiative, 2000)以来, 截至2020年底, 已有788个植物物种的1 031个基因组被测序和报道(Sun et al., 2022b)。其中农作物的基因组测序受到更多的关注, 已测序物种最多的禾本科(Poaceae)、十字花科(Brassicaceae)和豆科(Fabaceae)均为重要的经济作物科(Sun et al., 2022b)。虽然林木物种基因组研究快速发展, 已测序乔木物种达218种, 占已测序维管植物的29% (图1A) (谢玲娟等, 2021), 而许多木本植物的大科尚未得到足够重视。在全球最大的5个木本植物科中(Beech et al., 2017), 仅有桃金娘科(Myrtaceae)已测序24个乔木物种, 为已测序乔木最多的科, 豆科、茜草科(Rubiaceae)、樟科(Lauraceae)和大戟科(Euphorbiaceae)仅有几个代表物种完成测序(图1B)。
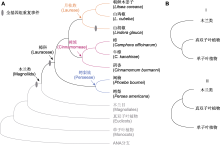
自2019年樟科第1个全基因组, 即牛樟(Cinnamomum kanehirae=Camphora kanahirae) (Chung and Hsieh, 2023)的基因组序列发表以来(Chaw et al., 2019), 樟科全基因组研究呈现蓬勃发展的态势。目前已有8个物种完成测序(图2; 表1), 分布于樟科3个族(3/9) 6个属(约6/50)。牛樟基因组不仅是樟科首个完成全基因组测序的物种, 也是木兰分支第1个测序的物种(Chaw et al., 2019)。该研究从全基因组层面探讨了木兰类与双子叶植物的关系, 发掘了与牛樟形成相关的基因。随后, 在2019-2020年, 樟科重要的水果鳄梨(Persea americana)、化学物质原料山苍子(Litsea cubeba)以及优良木材树种闽楠(Phoebe bournei)的基因组相继发表(Rendón-Anaya et al., 2019; Chen et al., 2020a, 2020b)。这些物种基因组丰富了樟科基因组数据库, 为樟科基因组研究奠定了基础。
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al., 2022a)。仅一年间共有9篇樟科基因组文章发表(图2C; 表1), 樟(Camphora officinarum) (Jiang et al., 2022; Shen et al., 2022; Sun et al., 2022a; Wang et al., 2022)、阴香(Cinnamomum burmanni) (Li et al., 2022)、朝鲜木姜子(Litsea coreana) (Zhang et al., 2022)及山胡椒(Lindera glauca) (Xiong et al., 2022)的全基因组序列相继发布, 鳄梨和闽楠的基因组质量得到提升(Han et al., 2022; Nath et al., 2022)。尤其值得关注的是, 樟作为我国重要的经济树种, 2022年有4个高质量基因组由不同单位主导发表(图2B; 表1)。
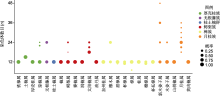
基因组大小及染色体数目是基因组的基本特征, 是研究科内多倍化和进化的基础。迄今为止, 樟科染色体数目的研究涵盖24个属(约24/50) 136个种(约136/ 3 000) (Oginuma and Tobe, 2006; Rice et al., 2015)。樟科物种多为二倍体, 通常n=12 (图3; 附录2)。染色体数目变化在樟科属内十分常见, 其中月桂属(Laurus)染色体n=18, 21, 24, 27, 30, 33, 36, 表现出丰富的变异。根据已发表的樟科基因组数据(包括全基因组测序的8个物种) (表1; 附录3), 樟科基因组大小为719 (樟)-2 982 Mb (月桂(Laurus nobilis))。月桂族的基因组明显大于樟族和鳄梨族(图4A)。
基因组大小、杂合度和重复序列比例等是判断基因组复杂程度的标准(高胜寒等, 2018)。复杂基因组在测序和组装过程中会面临更大的困难(高胜寒等, 2018)。已测序樟科物种均为二倍体(2n=24)木本植物(表1), 基因组大小介于719-2 092 Mb之间。其中山胡椒基因组是已测序樟科物种中最大的基因组, 约为其它月桂族测序物种的2倍(表1)。樟科物种的基因组杂合度较高(图4B), 除阴香的杂合度为0.7%外, 其余物种的杂合度均大于1%。Sun等(2022a)测序的樟基因组杂合度高达2.9%, 是已测序樟科基因组中杂合度最高的。樟科基因组重复序列比例为46.1%-76.8%, 在樟科基因组中变化较大(图4C)。月桂族重复序列占比最高, 超过60%, 其中山胡椒的重复序列占比最高, 为76.8%。长末端重复序列(long terminal repeat, LTR)为樟科基因组中最常见的转座子(transposable elements, TE)。LTR在樟科已发表基因组中占比为17.0%- 52.5% (图4D), 在鳄梨基因组中比例最低, 但在闽楠基因组中比例最高(Chen et al., 2020a)。
高杂合度和大量重复序列给樟科基因组测序组装带来了极大困难。最早测序的牛樟、鳄梨、山苍子和闽楠的基因组组装完整度较低(BUSCO组装完整度不超过90%)或者仅组装到Scaffold水平(表1)。二代与三代测序技术结合及生物信息技术的发展极大地提高了全基因组组装质量, 得樟科基因组组装完整度和注释基因数量大幅提高。结合Hi-C (highthroughput/resolution chromosome conformation capture)组装技术和BioNano光学图谱, 樟科基因组组装的染色体挂载率得到极大改善, 从此前的46.2%-94.6%提升至95%以上(表1)。
樟科基因组研究中鉴定出3次多倍化事件(图2A)。在樟科所有测序基因组中都检测到木兰目和樟目分化之前发生的1次WGD事件(约118-147 Ma) (Sun et al., 2022a; Han et al., 2022)以及已测序樟科物种共同祖先中发生的1次WGD事件(约76-95 Ma) (Shen et al., 2022; Han et al., 2022)。较近的樟科WGD事件与晚白垩纪早期樟科植物随着冈瓦纳大陆的分裂快速辐射分化的时间一致(Chanderbali et al., 2001)。WGD事件通过改善基本生理活动和初级代谢使植物对环境变化的适应性增强, 促进了早期樟科物种的快速辐射(Chen et al., 2020a; Jiang et al., 2022; Sun et al., 2022a)。值得注意的是, 山胡椒基因组在与相近的山苍子分化后(14.90-23.18 Ma)独自经历了1次多倍化事件, 与早中新世(23 Ma)月桂族快速辐射分化的时间一致(Qin et al., 2023)。月桂族为樟科中雌雄异株类群, 主要分布在亚洲热带和亚热带地区, 为亚热带常绿阔叶林的建群种(Li et al., 2004; Qin et al., 2023)。这次多倍化事件导致山胡椒具有比山苍子及其它樟科植物大将近1倍的基因组和更多的蛋白质编码基因(表1) (Xiong et al., 2022)。其中, 与萜烯合酶生物合成相关的基因家族发生显著扩张, 可能对山胡椒的生态适应性和生物适应性有重要作用(Pichersky and Raguso, 2018; Xiong et al., 2022), 有利于其应对早中新世东亚季风盛行引起的极端天气(Qin et al., 2023)。
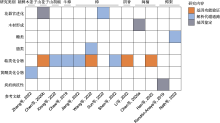
总体来说, 樟科基因组研究呈现3个明显的特点。(1) 樟科基因组特征信息十分匮乏。除樟族、月桂族和鳄梨族以外的樟科物种基因组大小未见报道(图2; 表1)。(2) 已测序物种明显地向中国分布及有经济价值的物种倾斜。首先, 测序物种主要集中在樟和闽楠等作为化学物质原料或提供优质木材的经济树种。其次, 2019年以来一直是我国学者在主导樟科基因组研究, 已发表的基因组中, 仅鳄梨基因组由国外学者报道, 其余全部为中国学者对国产种类的研究报道。此外, 已发表的樟科基因组集中在核心樟类群(core Lauraceae), 对樟科原始分支棠桂族、厚壳桂族、新樟族、檬果樟族及无根藤族等缺少研究(图2)。最后, 在空间分布上, 测序种类表现出明显的亚洲分布倾向, 作为樟科分布中心的热带非洲和南美洲物种(除鳄梨外)未有涉及, 这也与参与樟科基因组研究的中国学者居多有关。(3) 功能基因的挖掘不够深入。目前, 学者们侧重于挖掘重要的功能基因并解析其分子机制, 但樟科基因组研究还停留在基因层面, 缺少功能基因的验证及具体机制探究, 除TPS基因家族在樟科的起源与进化得到探讨外(Han et al., 2022), 对功能基因的系统研究和比较分析十分缺乏(图6)。此外, 与花器官进化、木材形成和代谢产物合成相关的功能基因是研究热点, 而樟科作为被子植物基部类群, 其经历漫长的历史变迁仍在森林中占据优势的独特生物学特性尚未得到足够的关注(图6)。
(2) 增加测序基因组分支的代表性。目前樟科测序的基因组仅涉及3个族, 还有6个族尚无代表物种的基因组发表(图2; 表1)。樟科具有复杂的进化历史, 从系统发生拓扑结构不一致中可窥探一二。有代表性的物种取样能够帮助厘清樟科的系统发生关系。注释良好的基因组数据为使用重测序和靶向捕获的方法从标本中获取大量的基因序列提供参考, 这将解决樟科采集和鉴定难题, 从而构建更具有代表性的系统发生树(Sun et al., 2022a)。一方面, 解析樟科系统发生关系不仅可帮助我们从进化角度探讨特定功能基因的生物学特性和分子机制, 还能为保护濒危植物提供理论依据。桫椤(Alsophila spinulosa)基因组就是1个良好的案例(Huang et al., 2022)。研究者基于9个群体107个桫椤个体的基因组重测序解析树蕨类植物树干形成发育的独特性, 为理解蕨类植物的进化提供了新视角, 也从影响桫椤种群变化的因素中拓展了保护工作思路。另一方面, 基于系统发生树和分布数据可揭示樟科的时空分布格局, 解析其类群多样分布类型的成因。例如, 泛热带分布的厚壳桂属和琼楠属(Beilschmiedia) (Chanderbali et al., 2001), 东亚-北美间断分布的檫木属(Sassafras) (Yang et al., 2022b; Qin et al., 2023), 亚洲-大洋洲分布的土楠属(Endiandra) (Li et al., 2020), 以及仅分布在亚洲的新樟属(Neocinnamomun)和仅分布在美洲的桂土楠群(Chanderbali et al., 2001; Liu et al., 2021)。此外, 大量遗传信息和代表性取样能够为解析樟科复杂的进化历史提供支持(Liu et al., 2021; Tian et al., 2021)。目前, 樟科科内的多倍化只在山胡椒基因组中检测到(图5), 考虑到月桂族多样的染色体变化及其更大的基因组(图3), 增加月桂族物种全基因组测序, 有助于理解多倍化在樟科进化中的作用。
(4) 对功能基因进行深入挖掘。樟科基因组功能基因的挖掘集中在与花器官进化、木材形成和代谢产物合成相关的基因, 且已挖掘的功能基因多数还未得到验证, 不利于基因的后续应用(图6)。解析与关键特征起源相关的功能基因和基因家族, 如落叶习性、单性花和寄生习性应受到更多关注。樟科因其特殊的系统发生位置, 在研究生物学特征的起源与进化上具有重要意义, 解析其关键特征起源的分子机制有助于揭开其作为原始类群至今繁盛的秘密。推动樟科重要物种高质量泛基因组研究将加深我们对其多样性的理解, 并发现与重要性状和环境适应相关的基因和变异, 为分子机制研究、品种选育和分子育种提供指导(Sun et al., 2022b; 郝晨路等, 2022)。以樟科重要的经济树种樟为例, 已发表的4个樟基因组在杂合度、编码基因数量和重复序列比例上具有明显差异(表1), 表现出个体间的遗传多样性(Jiang et al., 2022; Wang et al., 2022; Sun et al., 2022b; Shen et al., 2022)。泛基因组研究有助于我们理解种内个体间的差异, 进而更好地利用樟树资源。虽然目前泛基因组研究主要集中在农作物, 截至2021年末林木物种中仅苹果(Malus) (Wang et al., 2023b)和杨属(Populus) (Zhang et al., 2019)开展了泛基因组研究(郝晨路等, 2022)。但基因组规模大、复杂度高的林木物种泛基因组研究仍极具挑战性(Wang et al., 2023a)。随着技术的进步, 林木物种的泛基因组研究不仅能够促进对乔木起源的认识, 还能够解析森林建群种应对环境异质性的分子机制, 这对理解森林对环境变化的响应具有重要意义(郝晨路等, 2022; Wang et al., 2023a)。
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al.,
本文的其它图/表
|
 首页
首页