
植物学报 ›› 2020, Vol. 55 ›› Issue (6): 715-732.DOI: 10.11983/CBB20091 cstr: 32102.14.CBB20091
赵宇慧1, 李秀秀1,2, 陈倬1,2, 鲁宏伟1,2, 刘羽诚1,2, 张志方1,2, 梁承志1,2,*( )
)
收稿日期:2020-05-20
接受日期:2020-08-26
出版日期:2020-11-01
发布日期:2020-11-11
通讯作者:
梁承志
作者简介:*E-mail: cliang@genetics.ac.cn基金资助:
Yuhui Zhao1, Xiuxiu Li1,2, Zhuo Chen1,2, Hongwei Lu1,2, Yucheng Liu1,2, Zhifang Zhang1,2, Chengzhi Liang1,2,*()
Received:2020-05-20
Accepted:2020-08-26
Online:2020-11-01
Published:2020-11-11
Contact:
Chengzhi Liang
摘要: 全基因组关联分析(GWAS)是动植物复杂性状相关基因定位的常用手段。高通量基因分型技术的应用极大地推动了GWAS的发展。在植物中, 利用GWAS不仅能够以较高的分辨率在全基因组水平鉴定出各种自然群体特定性状相关的基因或区间, 而且可揭示表型变异的遗传架构全景图。目前, 人们利用GWAS分析方法已在拟南芥(Arabidopsis thaliana)、水稻(Oryza sativa)、小麦(Triticum aestivum)、玉米(Zea mays)和大豆(Glycine max)等模式植物和重要农作物品系中发掘出与各种性状显著相关的数量性状座位(QTL)及其候选基因位点, 阐明了这些性状的遗传基础, 并为揭示这些性状背后的分子机理提供候选基因, 也为作物高产优质品种的选育提供了理论依据。该文对GWAS的方法、影响因素及数据分析流程进行了详细描述, 以期为相关研究提供参考。
赵宇慧, 李秀秀, 陈倬, 鲁宏伟, 刘羽诚, 张志方, 梁承志. 生物信息学分析方法I: 全基因组关联分析概述. 植物学报, 2020, 55(6): 715-732.
Yuhui Zhao, Xiuxiu Li, Zhuo Chen, Hongwei Lu, Yucheng Liu, Zhifang Zhang, Chengzhi Liang. An Overview of Genome-wide Association Studies in Plants. Chinese Bulletin of Botany, 2020, 55(6): 715-732.
| Method | Population structure | Kinship | Precision | Characteristic | Computational speed | Statistical power | Application |
|---|---|---|---|---|---|---|---|
| Standard MLM | P | All markers | Low | High | >100 papers | ||
| GRAMMAR | P | Approximate method | Very fast | Intermediate | Barley (200) | ||
| EMMA | P | Exact method | Intermediate | Similar to Standard MLM | >100 papers | ||
| EMMAX | P | All markers | Approximate method | High marker densities | Fast | Similar to Standard MLM | >100 papers |
| CMLM | P | Large sample sizes | Better than Standard MLM | >100 papers | |||
| FaST-LMM | P | A subset of genetic markers | Exact method | Large sample sizes | Fast | Similar to Standard MLM | Rice (200?1500) |
| GEMMA | P | Exact method | Fast | Similar to Standard MLM | Arabidopsis thaliana (190-500) | ||
| ECMLM | P | Intermediate | Better than Standard MLM | Sorghum (250-350), soybean (200-400), wheat (250-300) | |||
| GRAMMAR- Gamma | P | Approximate method | High marker densities | Fast | Similar to Standard MLM | Oilseed rape (200) | |
| SUPER | P | Trait-associated markers | Large sample size & high marker density | Fast | Better than Standard MLM | Wheat (300-400) | |
| Farm-CPU | P | A subset of genetic markers | Approximate method | Large sample size & high marker density | Fast | Better than Standard MLM | Wheat (100-1200), maize (100-5000) |
| BLINK | P | A subset of genetic markers | Approximate method | Large sample size & high marker density | Faster than FarmCPU | Better than FarmCPU |
表1 不同混合线性模型(MLM)的性能比较
Table 1 Performance comparison of different methods in mixed linear model (MLM)
| Method | Population structure | Kinship | Precision | Characteristic | Computational speed | Statistical power | Application |
|---|---|---|---|---|---|---|---|
| Standard MLM | P | All markers | Low | High | >100 papers | ||
| GRAMMAR | P | Approximate method | Very fast | Intermediate | Barley (200) | ||
| EMMA | P | Exact method | Intermediate | Similar to Standard MLM | >100 papers | ||
| EMMAX | P | All markers | Approximate method | High marker densities | Fast | Similar to Standard MLM | >100 papers |
| CMLM | P | Large sample sizes | Better than Standard MLM | >100 papers | |||
| FaST-LMM | P | A subset of genetic markers | Exact method | Large sample sizes | Fast | Similar to Standard MLM | Rice (200?1500) |
| GEMMA | P | Exact method | Fast | Similar to Standard MLM | Arabidopsis thaliana (190-500) | ||
| ECMLM | P | Intermediate | Better than Standard MLM | Sorghum (250-350), soybean (200-400), wheat (250-300) | |||
| GRAMMAR- Gamma | P | Approximate method | High marker densities | Fast | Similar to Standard MLM | Oilseed rape (200) | |
| SUPER | P | Trait-associated markers | Large sample size & high marker density | Fast | Better than Standard MLM | Wheat (300-400) | |
| Farm-CPU | P | A subset of genetic markers | Approximate method | Large sample size & high marker density | Fast | Better than Standard MLM | Wheat (100-1200), maize (100-5000) |
| BLINK | P | A subset of genetic markers | Approximate method | Large sample size & high marker density | Faster than FarmCPU | Better than FarmCPU |
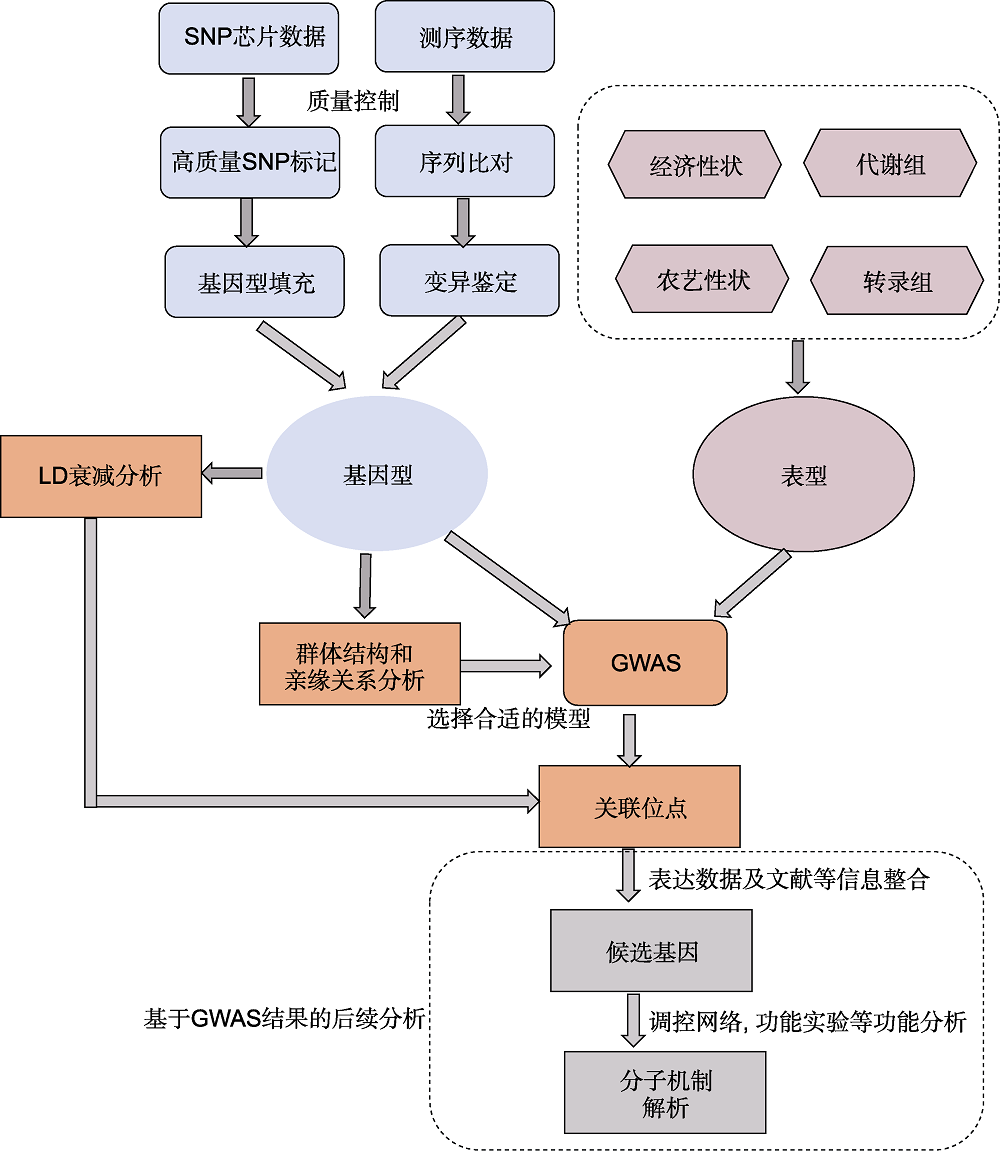
图1 全基因组关联分析(GWAS)流程
Figure 1 The pipeline of genome-wide association study (GWAS)
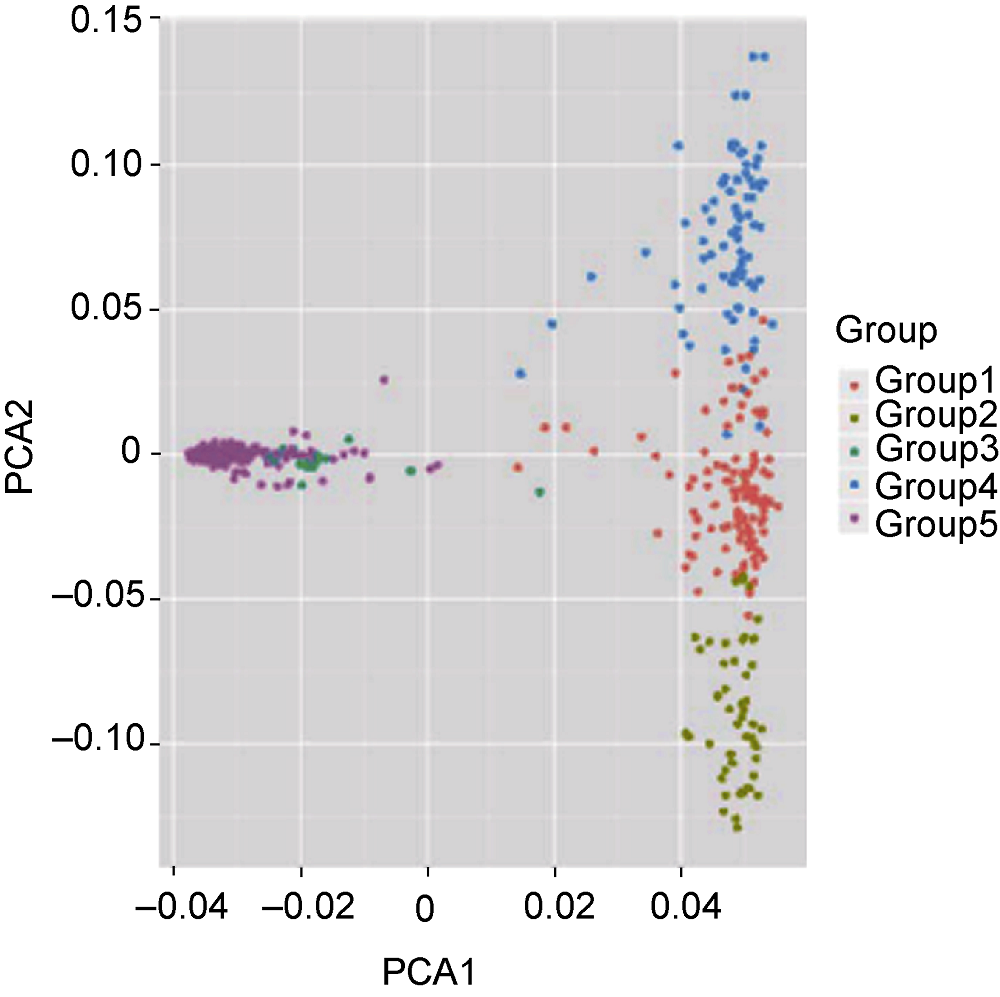
图2 721份水稻材料的主成分分析(PCA)图
Figure 2 The first two components from principal component analysis (PCA) of 721 rice accessions
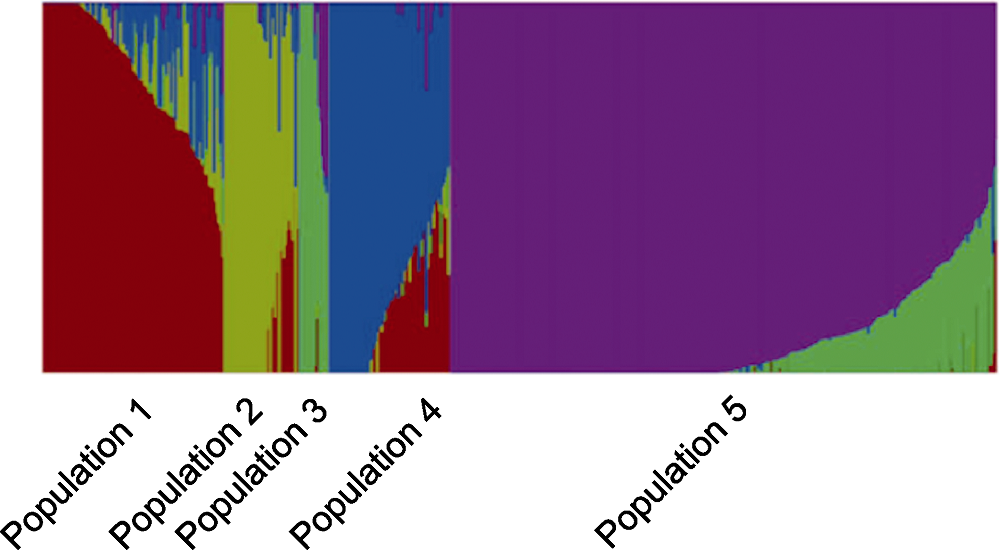
图3 721份水稻材料的群体结构分析
Figure 3 Population structure analyses of 721 rice accessions
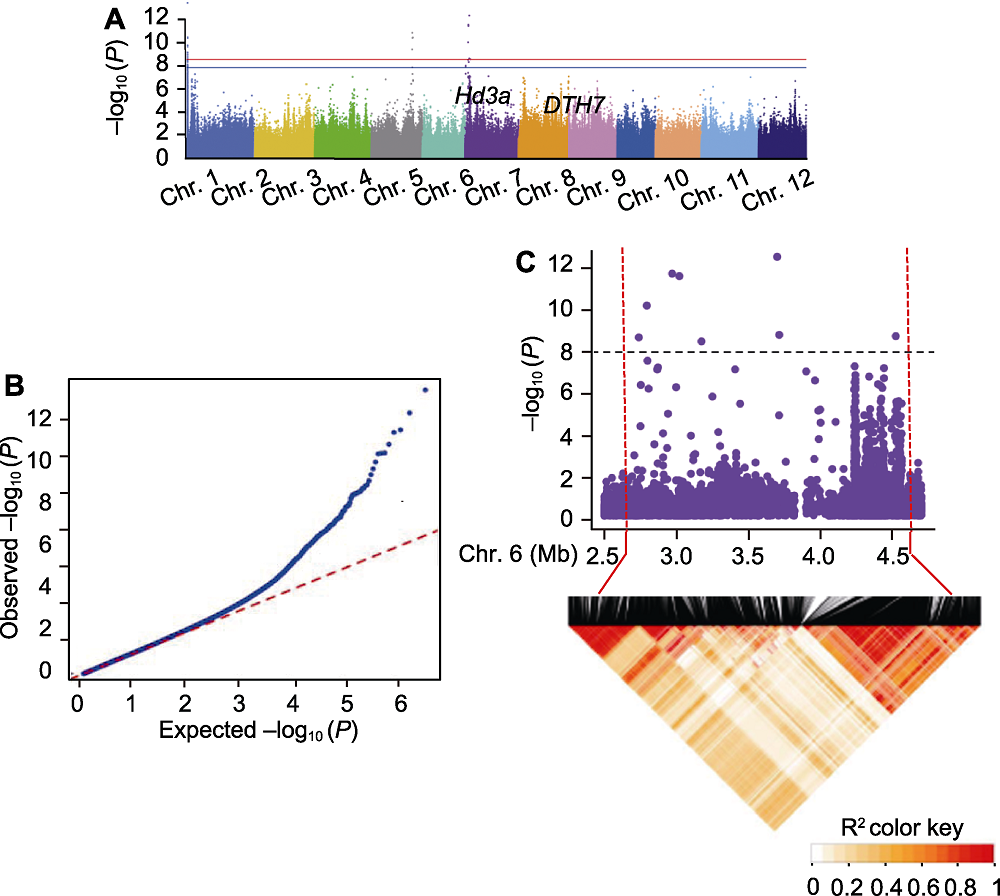
图4 721份水稻材料抽穗期全基因组关联分析(GWAS)结果展示 (A) 抽穗期性状关联分析结果的曼哈顿图; (B) QQ图; (C) 局部曼哈顿图和6号染色体尖峰附近的LD热图。曼哈顿图中红色虚线标出候选区间, 黑色虚线表示显著性阈值-log10 (P)=7.80。
Figure 4 Genome-wide association study (GWAS) results of 721 rice accessions for heading date (A) Manhattan plots of GWAS results for heading date; (B) QQ plot; (C) Local manhattan plots and LD heatmap around the peak on chromosome 6. Candidate region was labelled by red dotted line while the black dotted line indicated threshold -log10 (P)=7.80.
| [1] |
Abegaz F, Chaichoompu K, Génin E, Fardo DW, König IR, Mahachie John JM, Van Steen K (2019). Principals about principal components in statistical genetics. Brief Bioinform 20, 2200-2216.
DOI URL PMID |
| [2] |
Alexander DH, Novembre J, Lange K (2009). Fast model- based estimation of ancestry in unrelated individuals. Genome Res 19, 1655-1664.
DOI URL PMID |
| [3] |
Alqudah AM, Koppolu R, Wolde GM, Graner A, Schnurbusch T (2016). The genetic architecture of barley plant stature. Front Genet 7, 117.
DOI URL PMID |
| [4] |
Alqudah AM, Sallam A, Baenziger PS, Börner A (2020). GWAS: fast-forwarding gene identification and characterization in temperate cereals: lessons from barley—a review. J Adv Res 22, 119-135.
DOI URL PMID |
| [5] |
Aulchenko YS, de Koning DJ, Haley C (2007). Genomewide rapid association using mixed model and regression, a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics 177, 577-585.
DOI URL PMID |
| [6] | Bai XF, Zhao H, Huang Y, Xie WB, Han ZM, Zhang B, Guo ZL, Yang L, Dong HJ, Xue WY, Li GW, Hu G, Hu Y, Xing YZ (2016). Genome-wide association analysis reveals different genetic control in panicle architecture between Indica and Japonica rice. Plant Genome 9, 1-10. |
| [7] |
Barrett JC, Fry B, Maller J, Daly MJ (2005). Haploview, analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263-265.
DOI URL PMID |
| [8] |
Bhatta M, Baenziger PS, Waters BM, Poudel R, Belamkar V, Poland J, Morgounov A (2018). Genome-wide association study reveals novel genomic regions associated with 10 grain minerals in synthetic hexaploid wheat. Int J Mol Sci 19, 3237.
DOI URL |
| [9] |
Bolger AM, Lohse M, Usadel B (2014). Trimmomatic, a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114-2120.
DOI URL |
| [10] |
Bradbury PJ, Zhang ZW, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633-2635.
DOI URL PMID |
| [11] |
Casale FP, Rakitsch B, Lippert C, Stegle O (2015). Efficient set tests for the genetic analysis of correlated traits. Nat Methods 12, 755-758.
DOI URL PMID |
| [12] |
Chen J, Hu X, Shi TT, Yin HR, Sun DF, Hao YF, Xia XC, Luo J, Fernie AR, He ZH, Chen W (2020). Metabolite-based genome-wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnol J 18, 1722-1735.
DOI URL PMID |
| [13] |
Chen PL, Shen ZK, Ming LC, Li YB, Dan WH, Lou GM, Peng B, Wu B, Li YH, Zhao D, Gao GJ, Zhang QL, Xiao JH, Li XH, Wang GW, He YQ (2018). Genetic basis of variation in rice seed storage protein (Albumin, Globulin, Prolamin, and Glutelin) content revealed by genome-wide association analysis. Front Plant Sci 9, 612.
DOI URL PMID |
| [14] |
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group (2011). The variant call format and VCFtools. Bioinformatics 27, 2156-2158.
DOI URL |
| [15] | De R, Bush WS, Moore JH (2014). Bioinformatics challenges in genome-wide association studies (GWAS). In: Trent R, ed. Clinical Bioinformatics. Methods in Molecular Biology (Methods and Protocols), Vol. 1168. 1New York: Humana Press. pp. 63-81. |
| [16] |
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491-498.
DOI URL PMID |
| [17] |
Dong HJ, Zhao H, Li SL, Han ZM, Hu G, Liu C, Yang GY, Wang GW, Xie WB, Xing YZ (2018). Genome-wide association studies reveal that members of bHLH subfamily 16 share a conserved function in regulating flag leaf angle in rice ( Oryza sativa). PLoS Genet 14, e1007323.
DOI URL PMID |
| [18] |
Dong XK, Gao YQ, Chen W, Wang WS, Gong L, Liu XQ, Luo J (2015). Spatiotemporal distribution of phenolamides and the genetics of natural variation of hydroxycinnamoyl spermidine in rice. Mol Plant 8, 111-121.
URL PMID |
| [19] |
Du XM, Huang G, He SP, Yang ZE, Sun GF, Ma XF, Li N, Zhang XY, Sun JL, Liu M, Jia YH, Pan ZE, Gong WF, Liu ZH, Zhu HQ, Ma L, Liu FY, Yang DG, Wang F, Fan W, Gong Q, Peng Z, Wang LR, Wang XY, Xu SJ, Shang HH, Lu CR, Zheng HK, Huang SW, Lin T, Zhu YX, Li FG (2018). Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat Genet 50, 796-802.
DOI URL PMID |
| [20] |
Duan NB, Bai Y, Sun HH, Wang N, Ma YM, Li MJ, Wang X, Jiao C, Legall N, Mao LY, Wan SB, Wang K, He TM, Feng SQ, Zhang ZY, Mao ZQ, Shen X, Chen XL, Jiang YM, Wu SJ, Yin CM, Ge SF, Yang L, Jiang SH, Xu HF, Liu JX, Wang DY, Qu CZ, Wang YC, Zuo WF, Xiang L, Liu C, Zhang DY, Gao Y, Xu YM, Xu KN, Chao T, Fazio G, Shu HR, Zhong GY, Cheng LL, Fei ZJ, Chen XS (2017). Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nat Commun 8, 249.
DOI URL PMID |
| [21] |
Fabres PJ, Collins C, Cavagnaro TR, Rodríguez López CM (2017). A concise review on multi-omics data integration for terroir analysis in Vitis vinifera. Front Plant Sci 8, 1065.
URL PMID |
| [22] |
Falush D, Stephens M, Pritchard JK (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567-1587.
URL PMID |
| [23] |
Fang C, Ma YM, Wu SW, Liu Z, Wang Z, Yang R, Hu GH, Zhou ZK, Yu H, Zhang M, Pan Y, Zhou GA, Ren HX, Du WG, Yan HR, Wang YP, Han DZ, Shen YT, Liu SL, Liu TF, Zhang JX, Qin H, Yuan J, Yuan XH, Kong FJ, Liu BH, Li JY, Zhang ZW, Wang GD, Zhu BG, Tian ZX (2017). Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol 18, 161.
DOI URL PMID |
| [24] |
Furlotte NA, Eskin E (2015). Efficient multiple-trait association and estimation of genetic correlation using the matrix-variate linear mixed model. Genetics 200, 59-68.
URL PMID |
| [25] |
Griffith OL, Montgomery SB, Bernier B, Chu B, Kasaian K, Aerts S, Mahony S, Sleumer MC, Bilenky M, Haeussler M, Griffith M, Gallo SM, Giardine B, Hooghe B, Van Loo P, Blanco E, Ticoll A, Lithwick S, Portales-Casamar E, Donaldson IJ, Robertson G, Wadelius C, De Bleser P, Vlieghe D, Halfon MS, Wasserman W, Hardison R, Bergman CM, Jones SJM, Open Regulatory Annotation C (2008). ORegAnno: an open- access community-driven resource for regulatory annotation. Nucleic Acids Res 36, D107-D113.
DOI URL PMID |
| [26] |
Gumpinger AC, Roqueiro D, Grimm DG, Borgwardt KM (2018). Methods and tools in genome-wide association studies. Methods Mol Biol 1819, 93-136.
DOI URL PMID |
| [27] |
Guo SG, Zhao SJ, Sun HH, Wang X, Wu S, Lin T, Ren Y, Gao L, Deng Y, Zhang J, Lu XQ, Zhang HY, Shang JL, Gong GY, Wen CL, He N, Tian SW, Li MY, Liu JP, Wang YP, Zhu YC, Jarret R, Levi A, Zhang XP, Huang SW, Fei ZJ, Liu WG, Xu Y (2019). Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat Genet 51, 1616-1623.
URL PMID |
| [28] | Huang M, Liu XL, Zhou Y, Summers RM, Zhang ZW (2019). BLINK: a package for the next level of genome- wide association studies with both individuals and markers in the millions. Gigascience 8, giy154. 8, giy154. |
| [29] |
Huang XH, Yang SH, Gong JY, Zhao Y, Feng Q, Gao H, Li WJ, Zhan QL, Cheng BY, Xia JH, Chen N, Hao ZN, Liu KY, Zhu CR, Huang T, Zhao Q, Zhang L, Fan DL, Zhou CC, Lu YQ, Weng QJ, Wang ZX, Li JY, Han B (2015). Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat Commun 6, 6258.
DOI URL PMID |
| [30] |
Huang XH, Yang SH, Gong JY, Zhao Q, Feng Q, Zhan QL, Zhao Y, Li WJ, Cheng BY, Xia JH, Chen N, Huang T, Zhang L, Fan DL, Chen JY, Zhou CC, Lu YQ, Weng QJ, Han B (2016). Genomic architecture of heterosis for yield traits in rice. Nature 537, 629-633.
DOI URL PMID |
| [31] |
Huang XH, Zhao Y, Wei XH, Li CY, Wang AH, Zhao Q, Li WJ, Guo YL, Deng LW, Zhu CR, Fan DL, Lu YQ, Weng QJ, Liu KY, Zhou TY, Jing YF, Si LZ, Dong GJ, Huang T, Lu TT, Feng Q, Qian Q, Li JY, Han B (2011). Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet 44, 32-39.
DOI URL PMID |
| [32] |
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009). Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9, 1322-1332.
DOI URL PMID |
| [33] | Hübner S, Bercovich N, Todesco M, Mandel JR, Odenheimer J, Ziegler E, Lee JS, Baute GJ, Owens GL, Grassa CJ, Ebert DP, Ostevik KL, Moyers BT, Yakimowski S, Masalia RR, Gao LX, Ćalić I, Bowers JE, Kane NC, Swanevelder DZH, Kubach T, Muños S, Langlade NB, Burke JM, Rieseberg LH (2019). Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat Plants 5, 54-62. |
| [34] | Jiang D, Wang MY (2018). Recent developments in statistical methods for GWAS and high-throughput sequencing association studies of complex traits. Biostatist Epidemiol 2, 132-159. |
| [35] |
Jin ML, Liu HJ, He C, Fu JJ, Xiao YJ, Wang YB, Xie WB, Wang GY, Yan JB (2016). Maize pan-transcriptome provides novel insights into genome complexity and quantitative trait variation. Sci Rep 6, 18936.
DOI URL PMID |
| [36] |
Kaler AS, Purcell LC (2019). Estimation of a significance threshold for genome-wide association studies. BMC Genomics 20, 618.
DOI URL PMID |
| [37] | Kaler AS, Ray JD, Schapaugh WT, King CA, Purcell LC (2017). Genome-wide association mapping of canopy wilting in diverse soybean genotypes. Theor Appl Genet 130, 2203-2217. |
| [38] |
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, Sabatti C, Eskin E (2010). Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42, 348-354.
DOI URL PMID |
| [39] |
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E (2008). Efficient control of population structure in model organism association mapping. Genetics 178, 1709-1723.
DOI URL PMID |
| [40] |
Kidane YG, Gesesse CA, Hailemariam BN, Desta EA, Mengistu DK, Fadda C, Pè ME, Dell'Acqua M (2019). A large nested association mapping population for breeding and quantitative trait locus mapping in Ethiopian durum wheat. Plant Biotechnol J 17, 1380-1393.
DOI URL PMID |
| [41] |
Korte A, Vilhjálmsson BJ, Segura V, Platt A, Long Q, Nordborg M (2012). A mixed-model approach for genome-wide association studies of correlated traits in structured populations. Nat Genet 44, 1066-1071.
URL PMID |
| [42] |
Kremling KAG, Chen SY, Su MH, Lepak NK, Romay MC, Swarts KL, Lu F, Lorant A, Bradbury PJ, Buckler ES (2018). Dysregulation of expression correlates with rare-allele burden and fitness loss in maize. Nature 555, 520-523.
URL PMID |
| [43] |
Kremling KAG, Diepenbrock CH, Gore MA, Buckler ES, Bandillo NB (2019). Transcriptome-wide association supplements genome-wide association in Zea mays. G3 9, 3023-3033.
DOI URL PMID |
| [44] | Kumar J, Pratap A, Solanki RK, Gupta DS, Goyal A, Chaturvedi SK, Nadarajan N, Kumar S (2012). Genomic resources for improving food legume crops. J Agric Sci 150, 289-318. |
| [45] |
Kusmec A, Srinivasan S, Nettleton D, Schnable PS (2017). Distinct genetic architectures for phenotype means and plasticities in Zea mays. Nat Plants 3, 715-723.
DOI URL PMID |
| [46] |
Langmead B, Trapnell C, Pop M, Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25.
DOI URL PMID |
| [47] |
Lee TH, Guo H, Wang XY, Kim C, Paterson AH (2014). SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics 15, 162.
DOI URL PMID |
| [48] | Li CH, Sun BC, Li YX, Liu C, Wu X, Zhang DF, Shi YS, Song YC, Buckler ES, Zhang ZW, Wang TY, Li Y (2016). Numerous genetic loci identified for drought tolerance in the maize nested association mapping populations. BMC Genomics 17, 894. |
| [49] |
Li CH, Wu X, Li YX, Shi YS, Song YC, Zhang DF, Li Y, Wang TY (2019). Genetic architecture of phenotypic means and plasticities of kernel size and weight in maize. Theor Appl Genet 132, 3309-3320.
DOI URL PMID |
| [50] |
Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987-2993.
DOI URL PMID |
| [51] | Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: 1303. 3997. |
| [52] |
Li M, Liu XL, Bradbury P, Yu JM, Zhang YM, Todhunter RJ, Buckler ES, Zhang ZW (2014). Enrichment of statistical power for genome-wide association studies. BMC Biol 12, 73.
DOI URL |
| [53] |
Li XX, Chen Z, Zhang GM, Lu HW, Qin P, Qi M, Yu Y, Jiao BK, Zhao XF, Gao Q, Wang H, Wu YY, Ma JT, Zhang LY, Wang YL, Deng LW, Yao SG, Cheng ZK, Yu DQ, Zhu LH, Xue YB, Chu CC, Li AH, Li SG, Liang CZ (2020). Analysis of genetic architecture and favorable allele usage of agronomic traits in a large collection of Chinese rice accessions. Sci China Life Sci 63, 1688-1702.
DOI URL PMID |
| [54] |
Li YL, Ruperao P, Batley J, Edwards D, Davidson J, Hobson K, Sutton T (2017). Genome analysis identified novel candidate genes for Ascochyta blight resistance in chickpea using whole genome re-sequencing data. Front Plant Sci 8, 359.
DOI URL PMID |
| [55] |
Li YL, Ruperao P, Batley J, Edwards D, Khan T, Colmer TD, Pang JY, Siddique KHM, Sutton T (2018). Investigating drought tolerance in chickpea using genome-wide association mapping and genomic selection based on whole-genome resequencing data. Front Plant Sci 9, 190.
DOI URL PMID |
| [56] |
Lippert C, Listgarten J, Liu Y, Kadie CM, Davidson RI, Heckerman D (2011). FaST linear mixed models for genome-wide association studies. Nat Methods 8, 833-835.
URL PMID |
| [57] |
Listgarten J, Lippert C, Heckerman D (2013). FaST-LMM- Select for addressing confounding from spatial structure and rare variants. Nat Genet 45, 470-471.
URL PMID |
| [58] |
Liu HJ, Wang XQ, Warburton ML, Wen WW, Jin ML, Deng M, Liu J, Tong H, Pan QC, Yang XH, Yan JB (2015). Genomic, transcriptomic, and phenomic variation reveals the complex adaptation of modern maize breeding. Mol Plant 8, 871-884.
DOI URL PMID |
| [59] |
Liu HJ, Yan JB (2019). Crop genome-wide association study: a harvest of biological relevance. Plant J 97, 8-18.
URL PMID |
| [60] |
Liu XL, Huang M, Fan B, Buckler E, Zhang ZW (2016). Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet 12, e1005767.
DOI URL PMID |
| [61] |
Loh PR, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B, Patterson N, Price AL (2015). Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet 47, 284-290.
DOI URL PMID |
| [62] |
Lozada D, Godoy JV, Murray TD, Ward BP, Carter AH (2019). Genetic dissection of snow mold tolerance in US Pacific Northwest winter wheat through genome-wide association study and genomic selection. Front Plant Sci 10, 1337.
DOI URL PMID |
| [63] |
Ma HL, Li GL, W€urschum T, Zhang Y, Zheng DB, Yang XH, Li JS, Liu WX, Yan JB, Chen SJ (2018a). Genome-wide association study of haploid male fertility in maize ( Zea mays L.). Front Plant Sci 9, 974.
URL PMID |
| [64] |
Ma XF, Wang ZY, Li W, Zhang YZ, Zhou XJ, Liu YG, Ren ZY, Pei XY, Zhou KH, Zhang WS, He KL, Zhang F, Liu JF, Ma WY, Xiao GH, Yang DG (2019a). Resequencing core accessions of a pedigree identifies derivation of genomic segments and key agronomic trait loci during cotton improvement. Plant Biotechnol J 17, 762-775.
DOI URL PMID |
| [65] |
Ma XS, Feng FJ, Zhang Y, Elesawi IE, Xu K, Li TF, Mei HW, Liu HY, Gao NN, Chen CL, Luo LJ, Yu SW (2019b). A novel rice grain size gene OsSNB was identified by genome-wide association study in natural population. PLoS Genet 15, e1008191.
DOI URL PMID |
| [66] |
Ma ZY, He SP, Wang XF, Sun JL, Zhang Y, Zhang GY, Wu LQ, Li ZK, Liu ZH, Sun GF, Yan YY, Jia YH, Yang J, Pan ZE, Gu QS, Li XY, Sun ZW, Dai PH, Liu ZW, Gong WF, Wu JH, Wang M, Liu HW, Feng KY, Ke H, Wang JD, Lan HY, Wang GN, Peng J, Wang N, Wang LR, Pang BY, Peng Z, Li RQ, Tian SL, Du XM (2018b). Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat Genet 50, 803-813.
DOI URL PMID |
| [67] |
Martin ER, Tunc I, Liu Z, Slifer SH, Beecham AH, Beecham GW (2018). Properties of global- and local- ancestry adjustments in genetic association tests in admixed populations. Genet Epidemiol 42, 214-229.
DOI URL PMID |
| [68] |
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297-1303.
DOI URL PMID |
| [69] |
Milner SG, Jost M, Taketa S, Mazón ER, Himmelbach A, Oppermann M, Weise S, Knüpffer H, Basterrechea M, König P, Schüler D, Sharma R, Pasam RK, Rutten T, Guo GG, Xu DD, Zhang J, Herren G, Müller T, Krattinger SG, Keller B, Jiang Y, González MY, Zhao YS, Habekuß A, Färber S, Ordon F, Lange M, Börner A, Graner A, Reif JC, Scholz U, Mascher M, Stein N (2019). Genebank genomics highlights the diversity of a global barley collection. Nat Genet 51, 319-326.
DOI URL PMID |
| [70] |
Myles S, Peiffer J, Brown PJ, Ersoz ES, Zhang ZW, Costich DE, Buckler ES (2009). Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell 21, 2194-2202.
DOI URL PMID |
| [71] |
Peng YC, Liu HB, Chen J, Shi TT, Zhang C, Sun DF, He ZH, Hao YF, Chen W (2018). Genome-wide association studies of free amino acid levels by six multi-locus models in bread wheat. Front Plant Sci 9, 1196.
DOI URL |
| [72] |
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904-909.
DOI URL PMID |
| [73] |
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945-959.
URL PMID |
| [74] |
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC (2007). PLINK: a tool set for whole-ge- nome association and population-based linkage analyses. Am J Hum Genet 81, 559-575.
DOI URL PMID |
| [75] |
Raj A, Stephens M, Pritchard JK (2014). fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573-589.
DOI URL PMID |
| [76] |
Sallam A, Martsch R (2015). Association mapping for frost tolerance using multi-parent advanced generation inter-cross (MAGIC) population in faba bean ( Vicia faba L.). Genetica 143, 501-514.
DOI URL PMID |
| [77] |
Song CX, Li W, Pei XY, Liu YG, Ren ZY, He KL, Zhang F, Sun K, Zhou XJ, Ma XF, Yang DG (2019). Dissection of the genetic variation and candidate genes of lint percentage by a genome-wide association study in upland cotton. Theor Appl Genet 132, 1991-2002.
DOI URL PMID |
| [78] |
Svishcheva GR, Axenovich TI, Belonogova NM, van Duijn CM, Aulchenko YS (2012). Rapid variance components-based method for whole-genome association analysis. Nat Genet 44, 1166-1170.
DOI URL PMID |
| [79] |
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D (2019). Benefits and limitations of genome-wide association studies. Nat Rev Genet 20, 467-484.
DOI URL PMID |
| [80] |
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30, 2725-2729.
DOI URL PMID |
| [81] |
Tang H, Peng J, Wang P, Risch NJ (2005). Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol 28, 289-301.
DOI URL PMID |
| [82] | Tang Y, Liu XL, Wang JB, Li M, Wang QS, Tian F, Su ZB, Pan YC, Liu D, Lipka AE, Buckler ES, Zhang ZW (2016). GAPIT Version 2: an enhanced integrated tool for genomic association and prediction. Plant Genome 9, 1-9. |
| [83] |
Thudi M, Khan AW, Kumar V, Gaur PM, Katta K, Garg V, Roorkiwal M, Samineni S, Varshney RK (2016). Whole genome re-sequencing reveals genome-wide variations among parental lines of 16 mapping populations in chickpea ( Cicer arietinum L.). BMC Plant Biol 16, 10.
DOI URL PMID |
| [84] |
Tieman D, Zhu GT, Resende MFR Jr, Lin T, Nguyen C, Bies D, Rambla JL, Beltran KSO, Taylor M, Zhang B, Ikeda K, Liu ZY, Fisher J, Zemach I, Monforte A, Zamir D, Granell A, Kirst M, Huang SW, Klee H (2017). A chemical genetic roadmap to improved tomato flavor. Science 355, 391-394.
DOI URL PMID |
| [85] |
Tong W, Kim TS, Park YJ (2016). Rice chloroplast genome variation architecture and phylogenetic dissection in diverse Oryza species assessed by whole-genome resequencing. Rice 9, 57.
DOI URL PMID |
| [86] |
Varshney RK, Thudi M, Roorkiwal M, He WM, Upadhyaya HD, Yang W, Bajaj P, Cubry P, Rathore A, Jian JB, Doddamani D, Khan AW, Garg V, Chitikineni A, Xu DW, Gaur PM, Singh NP, Chaturvedi SK, Nadigatla GVPR, Krishnamurthy L, Dixit GP, Fikre A, Kimurto PK, Sreeman SM, Bharadwaj C, Tripathi S, Wang J, Lee SH, Edwards D, Polavarapu KKB, Penmetsa RV, Crossa J, Nguyen HT, Siddique KHM, Colmer TD, Sutton T, von Wettberg E, Vigouroux Y, Xu X, Liu X (2019). Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity, domestication and agronomic traits. Nat Genet 51, 857-864.
DOI URL PMID |
| [87] |
Visscher PM, Brown MA, McCarthy MI, Yang J (2012). Five years of GWAS discovery. Am J Hum Genet 90, 7-24.
DOI URL PMID |
| [88] |
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J (2017). 10 years of GWAS discovery, biology, function, and translation. Am J Hum Genet 101, 5-22.
DOI URL PMID |
| [89] |
Wang HR, Xu X, Vieira FG, Xiao YH, Li ZK, Wang J, Nielsen R, Chu CC (2016). The power of inbreeding: NGS-based GWAS of rice reveals convergent evolution during rice domestication. Mol Plant 9, 975-985.
DOI URL PMID |
| [90] |
Wang K, Li MY, Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164.
DOI URL PMID |
| [91] |
Wang MH, Cordell HJ, Van Steen K (2019). Statistical methods for genome-wide association studies. Semin Cancer Biol 55, 53-60.
DOI URL PMID |
| [92] |
Wang MY, Xu SZ (2019). Statistical power in genome-wide association studies and quantitative trait locus mapping. Heredity 123, 287-306.
URL PMID |
| [93] |
Wang Q, Tang JL, Han B, Huang XH (2020). Advances in genome-wide association studies of complex traits in rice. Theor Appl Genet 133, 1415-1425 .
DOI URL PMID |
| [94] |
Wang QS, Tian F, Pan YC, Buckler ES, Zhang ZW (2014). A SUPER powerful method for genome wide association study. PLoS One 9, e107684.
DOI URL PMID |
| [95] |
Wang QX, Xie WB, Xing HK, Yan J, Meng XZ, Li XL, Fu XK, Xu JY, Lian XM, Yu SB, Xing YZ, Wang GW (2015). Genetic architecture of natural variation in rice chlorophyll content revealed by a genome-wide association study. Mol Plant 8, 946-957.
DOI URL PMID |
| [96] |
Wei W, Mesquita ACO, de A. Figueiró A, Wu X, Manjunatha S, Wickland DP, Hudson ME, Juliatti FC, Clough SJ (2017). Genome-wide association mapping of resistance to a Brazilian isolate of Sclerotinia sclerotiorum in soybean genotypes mostly from Brazil. BMC Genomics 18, 849.
DOI URL PMID |
| [97] |
Wen WW, Li D, Li X, Gao YQ, Li WQ, Li HH, Liu J, Liu HJ, Chen W, Luo J, Yan JB (2014). Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat Commun 5, 3438.
DOI URL PMID |
| [98] |
Wu J, Wang LF, Fu JJ, Chen JB, Wei SH, Zhang SL, Zhang J, Tang YS, Chen ML, Zhu JF, Lei L, Geng QH, Liu CL, Wu L, Li XM, Wang XL, Wang Q, Wang ZL, Xing SL, Zhang HK, Blair MW, Wang SM (2020). Resequencing of 683 common bean genotypes identifies yield component trait associations across a north-south cline. Nat Genet 52, 118-125.
DOI URL PMID |
| [99] |
Wu S, Tohge T, Cuadros-Inostroza A, Tong H, Tenenboim H, Kooke R, Méret M, Keurentjes JB, Nikoloski Z, Fernie AR, Willmitzer L, Brotman Y (2018). Mapping the Arabidopsis metabolic landscape by untargeted metabolomics at different environmental conditions. Mol Plant 11, 118-134.
DOI URL PMID |
| [100] |
Xiao YJ, Liu HJ, Wu LJ, Warburton M, Yan JB (2017). Genome-wide association studies in maize: praise and stargaze. Mol Plant 10, 359-374.
DOI URL PMID |
| [101] |
Xie WB, Wang GW, Yuan M, Yao W, Lyu K, Zhao H, Yang M, Li PB, Zhang X, Yuan J, Wang QX, Liu F, Dong HX, Zhang LJ, Li XL, Meng XZ, Zhang W, Xiong LZ, He YQ, Wang SP, Yu SB, Xu CG, Luo J, Li XH, Xiao JH, Lian XM, Zhang QF (2015). Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proc Natl Acad Sci USA 112, E5411-E5419.
DOI URL PMID |
| [102] |
Yang N, Lu YL, Yang XH, Huang J, Zhou Y, Ali F, Wen WW, Liu J, Li JS, Yan JB (2014). Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet 10, e1004573.
DOI URL PMID |
| [103] |
Yano K, Yamamoto E, Aya K, Takeuchi H, Lo PC, Hu L, Yamasaki M, Yoshida S, Kitano H, Hirano K, Matsuoka M (2016). Genome-wide association study using whole- genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat Genet 48, 927-934.
DOI URL PMID |
| [104] |
Yu JM, Buckler ES (2006). Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17, 155-160.
DOI URL PMID |
| [105] |
Yu JM, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38, 203-208.
DOI URL PMID |
| [106] |
Zhang C, Dong SS, Xu JY, He WM, Yang TL (2019a). PopLDdecay, a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786-1788.
DOI URL PMID |
| [107] |
Zhang TF, Wu TT, Wang LW, Jiang BJ, Zhen CX, Yuan S, Hou WS, Wu CX, Han T, Sun S (2019b). A combined linkage and GWAS analysis identifies QTLs linked to soybean seed protein and oil content. Int J Mol Sci 20, 5915.
DOI URL |
| [108] |
Zhang X, Zhang H, Li LJ, Lan H, Ren ZY, Liu D, Wu L, Liu HL, Jaqueth J, Pan GT, Gao SB (2016). Characterizing the population structure and genetic diversity of maize breeding germplasm in Southwest China using genome- wide SNP markers. BMC Genomics 17, 697.
DOI URL PMID |
| [109] |
Zhang YM, Jia ZY, Dunwell JM (2019c). Editorial: the applications of new multi-locus GWAS methodologies in the genetic dissection of complex traits. Front Plant Sci 10, 100.
DOI URL PMID |
| [110] |
Zhang ZW, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu JM, Arnett DK, Ordovas JM, Buckler ES (2010). Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42, 355-360.
DOI URL PMID |
| [111] |
Zhou X, Stephens M (2014). Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods 11, 407-409.
DOI URL |
| [112] |
Zhou XY, Huang XH (2019). Genome-wide association studies in rice: how to solve the low power problems? Mol Plant 12, 10-12.
DOI URL PMID |
| [113] |
Zhou ZK, Jiang Y, Wang Z, Gou ZH, Lyu J, Li WY, Yu YJ, Shu LP, Zhao YJ, Ma YM, Fang C, Shen YT, Liu TF, Li CC, Li Q, Wu M, Wang M, Wu YS, Dong Y, Wan WT, Wang X, Ding ZL, Gao YD, Xiang H, Zhu BG, Lee SH, Wang W, Tian ZX (2015). Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol 33, 408-414.
DOI URL PMID |
| [114] |
Zhu GT, Wang SC, Huang ZJ, Zhang SB, Liao QG, Zhang CZ, Lin T, Qin M, Peng M, Yang CK, Cao X, Han X, Wang XX, van der Knaap E, Zhang ZH, Cui X, Klee H, Fernie AR, Luo J, Huang SW (2018). Rewiring of the fruit metabolome in tomato breeding. Cell 172, 249-261.
DOI URL PMID |
| [1] | 闫姿伶, 陈晓宇, 姚蒙. 基于环境DNA宏条形码的无脊椎动物多样性研究: 生物信息学流程比较与评估[J]. 生物多样性, 2026, 34(1): 25369-. |
| [2] | 陶小所, 姚晓华, 姚有华. 藜麦LOC110717159基因克隆、生物信息学分析及其在拟南芥过表达分析[J]. 植物学报, 2026, 61(1): 53-67. |
| [3] | 陈靖彧, 王文庆, 罗诗语, 杨路祥, 汪慧骏, 吴天宇, 朱乾坤. 岩白菜TCP基因家族的表达模式及代谢关联分析[J]. 植物学报, 2026, 61(1): 26-36. |
| [4] | 杨莉, 曲茜彤, 陈子航, 邹婷婷, 王全华, 王小丽. 菠菜AT-hook基因家族鉴定与表达谱分析[J]. 植物学报, 2025, 60(3): 377-392. |
| [5] | 徐聪, 张飞宇, 俞道远, 孙新, 张峰. 土壤动物的分子分类预测策略评估[J]. 生物多样性, 2022, 30(12): 22252-. |
| [6] | 韩璐, 杨菲, 吴应明, 牛云明, 曾祎明, 陈立欣. 晋西黄土区典型乔灌木短期水分利用效率对环境因子的响应[J]. 植物生态学报, 2021, 45(12): 1350-1364. |
| [7] | 李格,孟小庆,李宗芸,朱明库. 甘薯盐胁迫响应基因IbMYB3的表达特征及生物信息学分析[J]. 植物学报, 2020, 55(1): 38-48. |
| [8] | 程广前,贾克利,李娜,邓传良,李书粉,高武军. 石刁柏核质体DNA的生物信息学分析及染色体定位[J]. 植物学报, 2019, 54(3): 328-334. |
| [9] | 刘魏, 童永鳌, 白洁. 水稻雄配子体发育过程中tRNA片段的生物信息学分析[J]. 植物学报, 2018, 53(5): 625-633. |
| [10] | 贾乐东, 李施蒙, 许代香, 曲存民, 李加纳, 王瑞. 甘蓝型油菜BnMYB80基因的生物信息学分析[J]. 植物学报, 2016, 51(5): 620-630. |
| [11] | 程甜, 魏强, 李广林. 中粒咖啡萜类合成酶基因家族的生物信息学分析[J]. 植物学报, 2016, 51(2): 235-250. |
| [12] | 徐晓婷, 王志恒, DimitarDimitrov. 批量下载GenBank基因序列数据的新工具——NCBIminer[J]. 生物多样性, 2015, 23(4): 550-555. |
| [13] | 黄儒, 苍晶, 于晶, 卢宝伟, 刘丽杰, 王健飞, 郭人铭, 徐琛. 冬小麦小RNA高通量测序及生物信息学分析[J]. 植物学报, 2014, 49(1): 8-18. |
| [14] | 张贵慰, 曾珏, 郭维, 罗琼. 水稻AT-hook基因家族生物信息学分析[J]. 植物学报, 2014, 49(1): 49-62. |
| [15] | 孙欣, 高莹, 杨云锋. 环境微生物的宏基因组学研究新进展[J]. 生物多样性, 2013, 21(4): 393-400. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
 首页
首页