
植物学报 ›› 2023, Vol. 58 ›› Issue (2): 261-273.DOI: 10.11983/CBB22224 cstr: 32102.14.CBB22224
彭焕文1,2,3, 王伟1,2,3,*( )
)
收稿日期:2022-09-19
接受日期:2022-11-12
出版日期:2023-03-01
发布日期:2023-03-15
通讯作者:
*E-mail: 基金资助:
Huanwen Peng1,2,3, Wei Wang1,2,3,*()
Received:2022-09-19
Accepted:2022-11-12
Online:2023-03-01
Published:2023-03-15
Contact:
*E-mail: 摘要: 系统发生学是研究生物类群间进化关系的学科。随着测序技术、分析方法和计算能力的改进, 分子数据被广泛应用,促进了系统发生学的快速发展。系统发生树已成为生态学和比较生物学等研究领域的有力工具。然而, 许多研究在进行系统发生树构建时更侧重各种软件的使用, 一些基本原则或注意事项有时会被弱化甚至忽视。该文详细介绍了基于分子数据进行系统发生树构建的工作流程和基本方法, 包括类群取样、分子标记选择、序列比对、分区及模型选择、序列联合分析以及拓扑结构检验等关键步骤。此外, 该文还为系统发生树构建常用的3种方法(最大简约法、最大似然法和贝叶斯法)提供了相应的软件操作流程和运行命令, 以期为相关研究提供参考。
彭焕文, 王伟. 基于分子数据的系统发生树构建. 植物学报, 2023, 58(2): 261-273.
Huanwen Peng, Wei Wang. Phylogenetic Tree Reconstruction Based on Molecular Data. Chinese Bulletin of Botany, 2023, 58(2): 261-273.
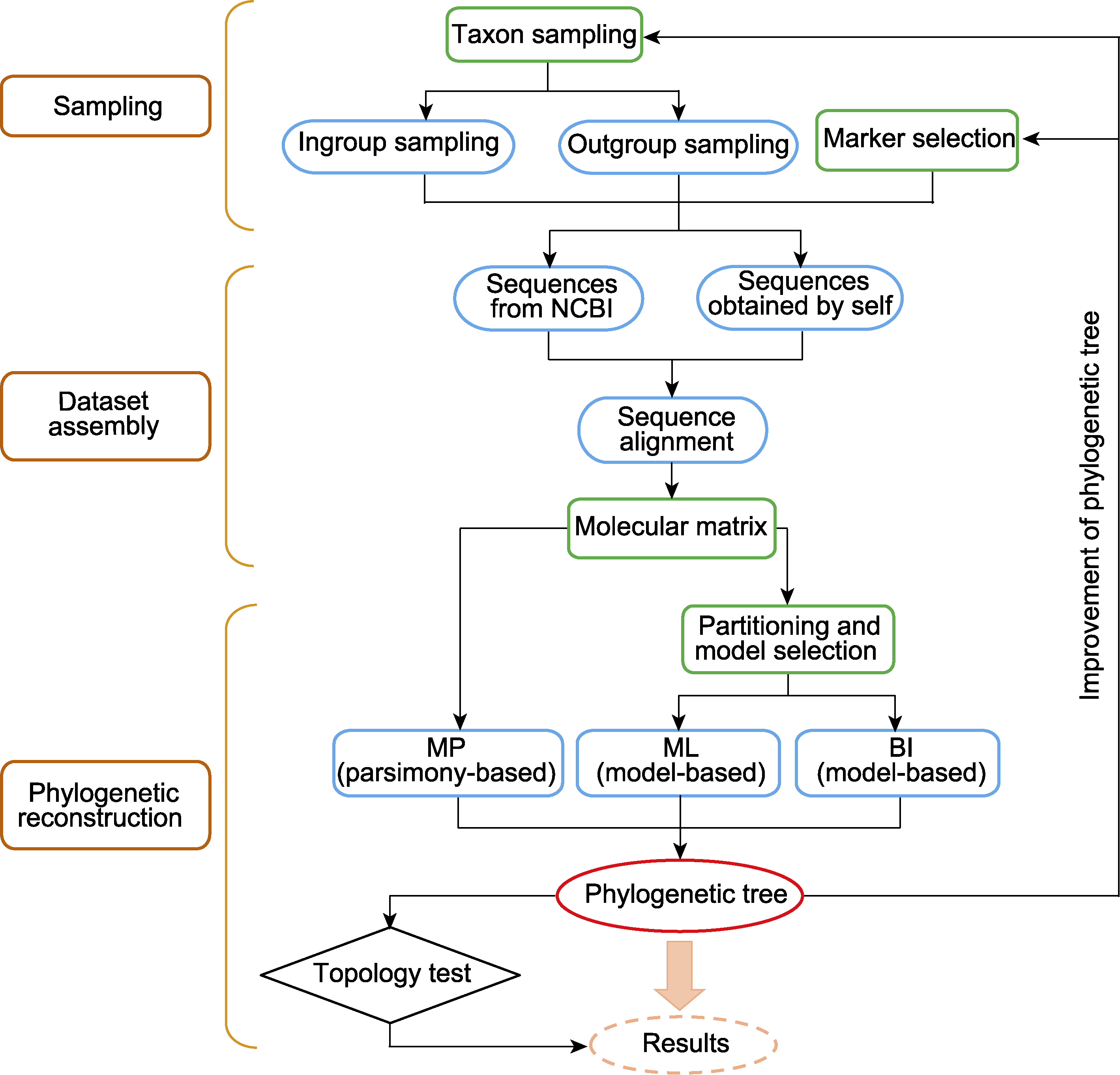
图1 基于分子数据构建系统发生树的流程 MP: 最大简约法; ML: 最大似然法; BI: 贝叶斯法
Figure 1 The pipeline of phylogenetic tree reconstruction based on molecular data MP: Maximum parsimony; ML: Maximum likelihood; BI: Bayesian inference

图2 Sanger测序获得的双向引物DNA序列色谱图 橙色方框示序列两端不明确碱基, 红色箭头示2个色谱图中相互冲突的碱基。
Figure 2 DNA sequence chromatograms obtained through Sanger sequencing using both forward and reverse primers Orange boxes show the ambiguous bases in both ends of the sequences, and red arrows show the conflicting base calls between two chromatograms.
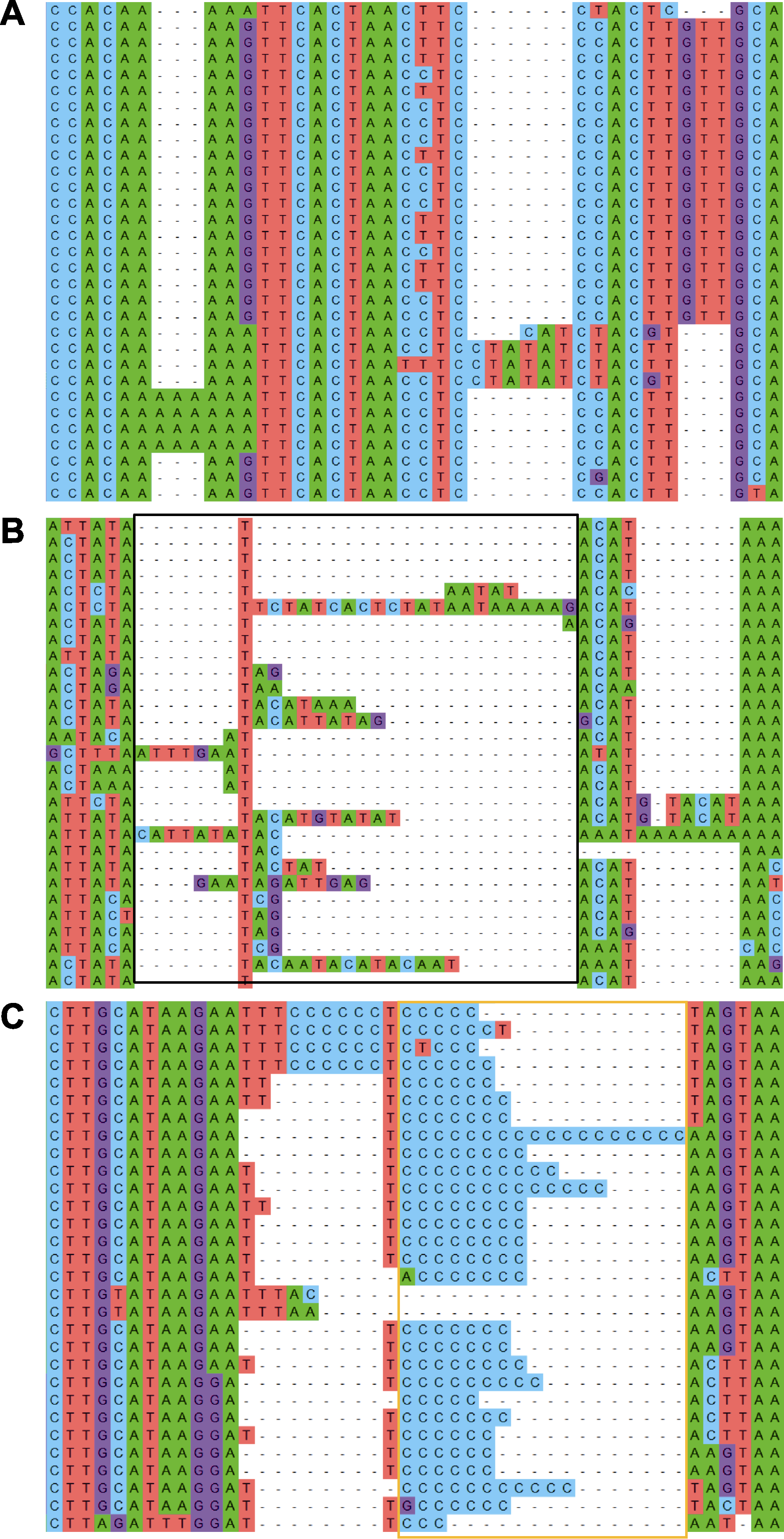
图3 序列比对示例 (A) 编码蛋白的序列比对; (B) 含有难排区域(黑色方框)的非编码序列比对; (C) 含有多聚碱基区域(橙色方框)的非编码序列比对
Figure 3 Examples for sequence alignment (A) Alignment for protein-coding sequences; (B) Alignment for non-coding sequences with a difficult-to-align region (indicated by black box); (C) Alignment for non-coding sequences with a poly-base region (indicated by orange box)
| [1] | 范凯, 叶方婷, 毛志君, 潘鑫峰, 李兆伟, 林文雄 (2021). 被子植物小热激蛋白家族的比较基因组学分析. 植物学报 56, 245-261. |
| [2] |
葛颂 (2022). 中国植物系统和进化生物学研究进展. 生物多样性 30, 22385.
DOI |
| [3] | 康凯程, 牛西强, 黄先忠, 胡能兵, 隋益虎, 张开京, 艾昊(2021). 辣椒R2R3-MYB转录因子家族的全基因组鉴定与比较进化分析. 植物学报 56, 315-329. |
| [4] | 王伟, 刘阳 (2020). 植物生命之树重建的现状、问题和对策建议. 生物多样性 28, 176-188. |
| [5] |
向小果, 王伟 (2015). 植物DNA条形码在系统发育研究中的应用. 生物多样性 23, 281-282.
DOI |
| [6] |
Benton MJ, Ayala FJ (2003). Dating the tree of life. Science 300, 1698-1700.
PMID |
| [7] |
Borsch T, Hilu KW, Quandt D, Wilde V, Neinhuis C, Barthlott W (2003). Noncoding plastid trnT-trnF sequences reveal a well resolved phylogeny of basal angiosperms. J Evol Biol 16, 558-576.
DOI URL |
| [8] |
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972-1973.
DOI PMID |
| [9] |
Castresana J (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17, 540-552.
DOI PMID |
| [10] |
Chase MW, Soltis DE, Olmstead RG, Morgan D, Les DH, Mishler BD, Duvall MR, Price RA, Hills HG, Qiu YL, Kron KA, Rettig JH, Conti E, Palmer JD, Manhart JR, Sytsma KJ, Michaels HJ, Kress WJ, Karol KG, Clark WD, Hedren M, Gaut BS, Jansen RK, Kim KJ, Wimpee CF, Smith JF, Furnier GR, Strauss SH, Xiang QY, Plunkett GM, Soltis PS, Swensen SM, Williams SE, Gadek PA, Quinn CJ, Eguiarte LE, Golenberg E, Learn Jr GH, Graham SW, Barrett SCH, Dayanandan S, Albert VA (1993). Phylogenetics of seed plants: an analysis of nucleotide sequences from the plastid gene rbcL. Ann Missouri Bot Gard 80, 528-580.
DOI URL |
| [11] |
Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792-1797.
DOI PMID |
| [12] |
Fan CZ, Xiang QY (2003). Phylogenetic analyses of Cornales based on 26S rRNA and combined 26S rDNA-matK- rbcL sequence data. Am J Bot 90, 1357-1372.
DOI URL |
| [13] |
Folk RA, Stubbs RL, Mort ME, Cellinese N, Allen JM, Soltis PS, Soltis DE, Guralnick RP (2019). Rates of niche and phenotype evolution lag behind diversification in a temperate radiation. Proc Natl Acad Sci USA 116, 10874-10882.
DOI PMID |
| [14] |
Goldman N, Anderson JP, Rodrigo AG (2000). Likelihood- based tests of topologies in phylogenetics. Syst Biol 49, 652-670.
PMID |
| [15] |
Goremykin VV, Nikiforova SV, Biggs PJ, Zhong BJ, Delange P, Martin W, Woetzel S, Atherton RA, Mclenachan PA, Lockhart PJ (2013). The evolutionary root of flowering plants. Syst Biol 62, 50-61.
DOI PMID |
| [16] | Hall TA (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/ 98/NT. Nucl Acids Symp Ser 41, 95-98. |
| [17] |
Jian SG, Soltis PS, Gitzendanner MA, Moore MJ, Li RQ, Hendry TA, Qiu YL, Dhingra A, Bell CD, Soltis DE (2008). Resolving an ancient, rapid radiation in Saxifragales. Syst Biol 57, 38-57.
DOI PMID |
| [18] | Joly S, McLenachan PA, Lockhart PJ (2009). A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am Nat 174, E54-E70. |
| [19] |
Kass RE, Raftery AE (1995). Bayes factors. J Am Stat Ass 90, 773-795.
DOI URL |
| [20] |
Katoh K, Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772-780.
DOI PMID |
| [21] |
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647-1649.
DOI PMID |
| [22] |
Kishino H, Hasegawa M (1989). Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol 29, 170-179.
PMID |
| [23] |
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34, 772-773.
DOI PMID |
| [24] |
Lian L, Ortiz RDC, Jabbour F, Chen ZD, Wang W (2019). Re-delimitation of Tinospora (Menispermaceae): implications for character evolution and historical biogeography. Taxon 68, 905-917.
DOI |
| [25] |
Liu GQ, Lian L, Wang W (2022). The molecular phylogeny of land plants: progress and future prospects. Diversity 14, 782.
DOI URL |
| [26] |
Liu L, Wu SY, Yu LL (2015). Coalescent methods for estimating species trees from phylogenomic data. J Syst Evol 53, 380-390.
DOI URL |
| [27] | Lozano-Fernandez J (2022). A practical guide to design and assess a phylogenomic study. Genome Biol Evol 14, evac129. |
| [28] |
Lu LM, Cox JC, Mathews S, Wang W, Wen J, Chen ZD (2018). Optimal data partitioning, multispecies coalescent and Bayesian concordance analyses resolve early divergences of the grape family (Vitaceae). Cladistics 34, 57-77.
DOI PMID |
| [29] |
Mirarab S, Nakhleh L, Warnow T (2021). Multispecies coalescent: theory and applications in phylogenetics. Annu Rev Ecol Evol Syst 52, 247-268.
DOI URL |
| [30] |
Nandi OI, Chase MW, Endress PK (1998). A combined cladistic analysis of angiosperms using rbcL and non-molecular data sets. Ann Missouri Bot Gard 85, 137-214.
DOI URL |
| [31] |
Nei M (1996). Phylogenetic analysis in molecular evolutio-a) nary genetics. Annu Rev Genet 30, 371-403.
PMID |
| [32] |
Owen CL, Marshall DC, Wade EJ, Meister R, Goemans G, Kunte K, Moulds M, Hill K, Villet M, Pham TH, Kortyna M, Lemmon EM, Lemmon AR, Simon C (2022). Detecting and removing sample contamination in phylogenomic data: an example and its implications for Cicadidae phylogeny (Insecta: Hemiptera). Syst Biol 71, 1504-1523.
DOI PMID |
| [33] |
Pelser PB, Kennedy AH, Tepe EJ, Shidler JB, Nordenstam B, Kadereit JW, Watson LE (2010). Patterns and causes of incongruence between plastid and nuclear Senecioneae (Asteraceae) phylogenies. Am J Bot 97, 856-873.
DOI PMID |
| [34] |
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol 67, 901-904.
DOI PMID |
| [35] |
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61, 539-542.
DOI PMID |
| [36] |
Schmidt HA, Strimmer K, Vingron M, von Haeseler A (2002). TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18, 502-504.
DOI PMID |
| [37] |
Shimodaira H (2002). An approximately unbiased test of phylogenetic tree selection. Syst Biol 51, 492-508.
DOI PMID |
| [38] |
Shimodaira H, Hasegawa M (1999). Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol 16, 1114.
DOI URL |
| [39] |
Shimodaira H, Hasegawa M (2001). CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246-1247.
PMID |
| [40] |
Smith MR (2013). Likelihood and parsimony diverge at high taxonomic levels. Cladistics 29, 463.
DOI PMID |
| [41] |
Soltis DE, Moore MJ, Burleigh G, Soltis PS (2009). Molecular markers and concepts of plant evolutionary relationships: progress, promise, and future prospects. Crit Rev Plant Sci 28, 1-15.
DOI URL |
| [42] |
Soltis PS, Soltis DE, Chase MW (1999). Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature 402, 402-404.
DOI |
| [43] |
Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312-1313.
DOI PMID |
| [44] |
Sun M, Folk RA, Gitzendanner MA, Soltis PS, Chen ZD, Soltis DE, Guralnick RP (2020). Recent accelerated diversification in rosids occurred outside the tropics. Nat Commun 11, 3333.
DOI PMID |
| [45] | Swofford DL (2002). PAUP*. Phylogenetic analysis using parsimony (* and other methods). Version 4. Sunderland, Massachusetts: Sinauer Associates. |
| [46] |
Tamura K, Stecher G, Kumar S (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38, 3022-3027.
DOI PMID |
| [47] |
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25, 4876-4882.
DOI PMID |
| [48] | van der Niet T, Linder HP (2008). Dealing with incongruence in the quest for the species tree: a case study from the orchid genus Satyrium. Mol Phylogenet Evol 47, 154-174. |
| [49] | Wang W (2018). A primer to the use of herbarium specimens in plant phylogenetics. Bot Lett 165, 404-408. |
| [50] |
Wang W, Del Ortiz RC, Jacques FMB, Chung SW, Liu Y, Xiang XG, Chen ZD (2017). New insights into the phylogeny of Burasaieae (Menispermaceae) with the recognition of a new genus and emphasis on the southern Taiwanese and mainland Chinese disjunction. Mol Phylogenet Evol 109, 11-20.
DOI PMID |
| [51] |
Wang W, Del Ortiz RC, Jacques FMB, Xiang XG, Li HL, Lin L, Li RQ, Liu Y, Soltis PS, Soltis DE, Chen ZD (2012). Menispermaceae and the diversification of tropical rainforests near the Cretaceous-Paleogene boundary. New Phytol 195, 470-478.
DOI PMID |
| [52] |
Wang W, Li HL, Chen ZD (2014a). Analysis of plastid and nuclear DNA data in plant phylogenetics—evaluation and improvement. Sci China Life Sci 57, 280-286.
DOI URL |
| [53] |
Wang W, Li HL, Xiang XG, Chen ZD (2014b). Revisiting the phylogeny of Ranunculeae: implications for divergence time estimation and historical biogeography. J Syst Evol 52, 551-565.
DOI URL |
| [54] |
Wang W, Wang HC, Chen ZD (2007). Phylogeny and morphological evolution of tribe Menispermeae (Menispermaceae) inferred from chloroplast and nuclear sequences. Perspect Plant Ecol Evol Syst 8, 141-154.
DOI URL |
| [55] |
Whelan S, Liò P, Goldman N (2001). Molecular phylogenetics: state-of-the-art methods for looking into the past. Trends Genet 17, 262-272.
DOI PMID |
| [56] |
Xi ZX, Ruhfel BR, Schaefer H, Amorim AM, Sugumaran M, Wurdack KJ, Endress PK, Matthews ML, Stevens PF, Mathews S, Davis CC (2012). Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proc Natl Acad Sci USA 109, 17519-17524.
DOI PMID |
| [57] | Xiang QY, Manchester SR, Thomas DT, Zhang WH, Fan CZ (2005). Phylogeny, biogeography, and molecular dating of cornelian cherries (Cornus, Cornaceae): tracking Tertiary plant migration. Evolution 59, 1685-1700. |
| [58] |
Yang ZH, Rannala B (2012). Molecular phylogenetics: principles and practice. Nat Rev Genet 13, 303-314.
DOI PMID |
| [1] | 任晓童, 张冉冉, 魏绍巍, 罗晓峰, 徐佳慧, 舒凯. 种子际微生物研究展望[J]. 植物学报, 2023, 58(3): 499-509. |
| [2] | 王伟, 刘阳. 植物生命之树重建的现状、问题和对策建议[J]. 生物多样性, 2020, 28(2): 176-188. |
| [3] | 陈作艺, 许晓静, 朱素英, 翟梦怡, 李扬. 中国沿海洛氏角毛藻复合群的多样性组成及地理分布[J]. 生物多样性, 2019, 27(2): 149-158. |
| [4] | 罗俊杰, 王莹, 商辉, 周喜乐, 韦宏金, 黄素楠, 顾钰峰, 金冬梅, 戴锡玲, 严岳鸿. 基于孢子形态和分子证据探讨鳞盖蕨属(碗蕨科)系统分类[J]. 植物学报, 2018, 53(6): 782-792. |
| [5] | 王伟, 张晓霞, 陈之端, 路安民. 被子植物APG分类系统评论[J]. 生物多样性, 2017, 25(4): 418-426. |
| [6] | 孙航, 邓涛, 陈永生, 周卓. 植物区系地理研究现状及发展趋势[J]. 生物多样性, 2017, 25(2): 111-122. |
| [7] | 杜周和, 刘俊凤, 刘彬斌, 左艳春, 吴建梅, 陈义安, 张剑飞, 鲁成. 家蚕地方品种遗传多样性及其分子系统学研究[J]. 生物多样性, 2013, 21(3): 315-325. |
| [8] | 黄继红, 张金龙, 杨永, 马克平. 特有植物多样性分布格局测度方法的新进展[J]. 生物多样性, 2013, 21(1): 99-110. |
| [9] | 李姝婧, 贾渝, 王庆华. 基部藓类分子系统学研究[J]. 植物学报, 2012, 47(4): 379-394. |
| [10] | 刘红梅, 张宪春, 曾辉, . DNA 序列在蕨类分子系统学研究中的应用[J]. 植物学报, 2009, 44(02): 143-158. |
| [11] | 姚戈 谢树莲. 串珠藻目分子系统学研究进展[J]. 植物学报, 2007, 24(02): 141-146. |
| [12] | 陆露 王红 李德铢. 杜鹃花科白珠树属分子系统学和生物地理学研究进展[J]. 植物学报, 2005, 22(06): 658-667. |
| [13] | 李晶 沙伟. 苔藓植物分子系统学研究概况[J]. 植物学报, 2004, 21(02): 172-179. |
| [14] | 李中奎 胡鸿钧 李夜光. 团藻目分子系统学研究进展[J]. 植物学报, 2002, 19(04): 419-424. |
| [15] | 王文. 分子系统学在生物保护中的意义[J]. 生物多样性, 1998, 06(2): 138-142. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
 首页
首页