
 首页
首页

全球气候变化导致生态系统物种多样性和功能多样性下降, 如何应对气候变化直接关系到生态系统的稳定性及其服务功能(Yuan et al., 2023; Jing et al., 2024)。植物如何应对气候变化是其持久生存的基础, 可通过局部适应或迁移到新的合适地点, 亦或通过表型可塑性适应气候变化(Alsos et al., 2012; Keenan, 2015; Poupon et al., 2021; Yang et al., 2022c)。然而, 这些应对策略效果有限, 气候的快速变化打破了基因与环境之间原有的联系, 导致迁徙、局部适应或新突变速率等无法与持续的气候变化保持平衡(Jia et al., 2020; Sang et al., 2022)。遗传多样性决定了物种适应新环境的能力, 也是生物进化的基础, 高杂合性可以抵消有害突变的影响, 增强物种的适应性和抗逆性(Pauls et al., 2013; Exposito-Alonso et al., 2022)。然而, 剧烈的气候变化导致物种遗传多样性大幅降低, 同时降低物种的持久性和进化潜力, 以及改变居群的遗传结构(Guan et al., 2021; Beridze et al., 2023; Liu et al., 2024)。景观遗传学(landscape genetics)为理解物种适应性进化和影响遗传变异的潜在环境因素提供了新见解(Alvarado et al., 2022; Feng and Du, 2022)。景观遗传学研究无需进行大量的胁迫实验, 仅通过遗传变异信息与环境信息或地理信息相结合就能揭示景观特征对遗传变异的影响(Aguirre-Liguori et al., 2021; Haupt and Schmid, 2022)。基因-环境关联(genotype-environment association, GEA)分析和异常位点检验(outlier tests)是了解遗传变异对整个景观适应模式的2种重要方法, 通过上述分析可以确定来自不同环境的物种遗传变异信息中参与环境适应的候选基因和基因组区域(Filipe et al., 2022; Haupt and Schmid, 2022)。利用景观遗传学方法揭示物种在全球环境变化条件下的适应机制, 已在动植物中广泛运用, 如三色黑鹂(Agelaius trico-lor)、小黄鱼(Larimichthys polyactis)、星叶草(Circaeaster agrestis)、茴芹叶茄(Solanum pimpinellifolium)和筒瓣花(Embothrium coccineum) (Gibson and Moyle, 2020; Zhang et al., 2020; Barr et al., 2021; Sepúlveda-Espinoza et al., 2022; Wang et al., 2022b)。
竹类遗传多样性研究已取得诸多成果, 但多数研究仅采用传统的分子标记, 如扩增片段长度多态性(amplified fragment length polymorphism, AFLP)、相关序列扩增多态性(sequence-related amplified poly-morphism, SRAP)、表达序列标签-微卫星标记(expressed sequence tags-simple sequence repeat, EST-SSR)、简单重复序列间扩增(inter-simple sequence repeat, ISSR)、简单重复序列(simple sequence repeat, SSR)、随机扩增多态性DNA (random amplified polymorphic DNA, RAPD)和序列标记微卫星(sequence-tagged microsatellite, STMS) (李潞滨等, 2008; Tian et al., 2012; Zhu et al., 2014; Bhandawat et al., 2019; Ely et al., 2019; Silva et al., 2020; Meena et al., 2023a)。随着测序技术的发展, 利用高通量测序可在居群水平上对全基因组范围内的变异信息进行筛选并提供大量的变异信息, 如单核苷酸多态性(single nucleotide polymorphism, SNP) (Shafer et al., 2015; Wu et al., 2023)。简化基因组测序(reduced-representation genome sequencing, RRGS)技术作为一种高效标记逐渐发展起来(Davey et al., 2011), 其中限制性酶切位点测序(restrictionsite associated DNA sequencing, RAD-seq)作为简化基因组测序, 可获得全基因组范围内的大量SNP位点, 且所产生的SNP位点在覆盖率、数据质量、稳定性和准确率方面比传统分子标记高, 适用于群体遗传学和景观基因组学研究(Morin et al., 2004; Baird et al., 2008; Ouborg et al., 2010; Lin et al., 2021)。RAD- seq技术在评估遗传多样性方面应用广泛, 特别是应用于珍稀濒危植物, 如荷叶铁线蕨(Adiantum nelumboides)、显脉木兰(Magnolia fistulosa)、海南风吹楠(Horsfieldia hainanensis)和云南蓝果树(Nyssa yunnanensis) (张珊珊等, 2019; 蔡超男等, 2021; 孙维悦等, 2022; Yang et al., 2022a)。目前, RAD-seq技术已应用于澜沧梨藤竹(Melocalamus arrectus)和云南省极小种群独龙江空竹(Cephalostachyum mannii)等竹类的遗传多样性评估(赵虎刚等, 2023; 张如礼等, 2024)。
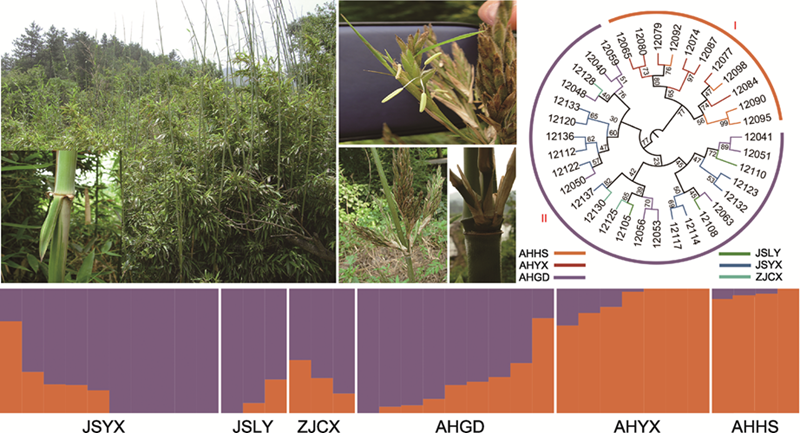
短穗竹(Brachystachyum densiflorum)为竹亚科(Bambusoideae)短穗竹属(Brachystachyum)植物, 是我国特有种, 其地下茎为细型, 亦称真鞭, 每节有3-5个分枝, 箨耳发达, 箨片开展易落, 假花序, 花枝极短缩, 呈短穗状或头状, 主要分布于华东地区(Li et al., 2006)。研究表明, 短穗竹可能由苦竹属(Pleioblastus)和刚竹属(Phyllostachys)杂交形成, 其分枝样式与苦竹属相似, 箨耳、花序和假小穗与刚竹属类似(Zhang et al., 2012)。近年来, 随着经济的快速发展, 城市化进程加快, 污染物排放量逐年增加(董芳淑等, 2023), 短穗竹的栖息地遭到破坏, 破碎化加剧, 种群数量呈减少趋势。目前, 短穗竹已被列入安徽省重点保护野生植物名录(
本研究针对中国特有种短穗竹, 利用RAD-seq技术获得了短穗竹6个居群的SNP数据集, 进而评估短穗竹居群遗传结构和遗传多样性水平, 同时基于景观遗传学方法, 分析气候变化对短穗竹现有遗传结构分布格局的影响和驱动因素, 利用物种分布模型(species distribution model, SDM)预测短穗竹当前潜在适生区以及未来气候变化下的分布区变化, 以期为短穗竹的保护提供理论依据。
1 材料与方法
1.1 植物材料采集
2012年6月在安徽、江苏和浙江采集到短穗竹(Brachystachyum densiflorum (Reudle) Keng) 6个居群的36个个体(表1), 每个个体间隔1 km以上, 以避免采集到来自同一个克隆的植株。每个居群采集3-10个个体, 每个个体采集3-5片幼嫩无病虫害的叶片, 置于分子采集袋中并迅速用硅胶干燥, 用于后续DNA提取。
表1 短穗竹居群采集信息
Table 1
| Population | Locality | Sample individuals | Longitude | Latitude |
|---|---|---|---|---|
| AHYX | Yuexi, Anhui | 7 | 116°14′49″ E | 30°55′27″ N |
| AHHS | Huoshan, Anhui | 4 | 116°26′38″ E | 31°24′01″ N |
| AHGD | Guangde, Anhui | 9 | 119°14′10″ E | 30°48′56″ N |
| JSLY | Liyang, Jiangsu | 3 | 119°28′30″ E | 31°14′39″ N |
| JSYX | Yixing, Jiangsu | 10 | 119°47′54″ E | 31°16′49″ N |
| ZJCX | Changxing, Zhejiang | 3 | 119°52′31″ E | 31°07′24″ N |
1.2 DNA提取、文库构建及测序
采用CTAB法(Doyle and Doyle, 1987)提取短穗竹全基因组DNA, 用1%琼脂糖凝胶电泳检测DNA提取质量, 确保符合后续建库要求。RAD建库采用单酶切法, 全基因组DNA用EcoRI (5'-GAATTC-3')限制性内切酶进行酶切, 在DNA片段的两端添加P1接头, 用超声波将其打断, 然后选择含有P1接头的片段添加P2接头, 并进行PCR扩增。将检测合格的PCR产物送至北京诺禾致源生物信息科技有限公司Illumina Novaseq 6000平台进行双端测序(PE=150 bp)。测序之后用Fastp v0.23.4 (
1.3 SNP筛选和过滤
以毛竹(Phyllostachys edulis)基因组(Zhao et al., 2018)为参考基因组。首先, 使用BWA v0.7.17软件(
1.4 遗传多样性和居群遗传结构分析
利用Stacks软件populations模块计算遗传多样性指标, 包括多态性信息含量(PIC)、观测杂合度(Ho)、期望杂合度(He)、核苷酸多样性(π)和近交系数(Fis)。使用NeEstimator v2.0软件(
1.5 遗传变异与气候关联分析
从WorldClim v.2.1数据库(Fick and Hijmans, 2017;
1.6 潜在地理分布预测
物种分布模型广泛用于预测当前和未来气候变化背景下物种的潜在分布(Yang et al., 2022b)。为明确过去和当前气候条件下短穗竹的潜在分布区以及预测未来全球气候变化下其空间分布趋势, 我们根据短穗竹已知位置进行物种分布建模。通过野外调查和检索全球生物多样性信息组织(
2 结果与分析
2.1 SNP位点统计和遗传多样性评估
研究显示, 经VCFtools软件过滤后保留了16 583个高质量SNPs (附图1), 其中9 196个SNPs发生了转换, 转换率为55.5%, 7 387个SNPs发生了颠换, 颠换率为44.5%, 转换与颠换的比值为1.24。基于16 583个SNPs评估6个短穗竹居群的遗传多样性(表2), 表明短穗竹居群具有中等水平遗传多样性(PIC=0.722 5, Ho=0.087, He=0.284 3, π=0.317 5), 各居群遗传多样性水平差异不大, 安徽广德(AHGD)和江苏宜兴(JSYX)两居群的遗传多样性较高, 而浙江长兴(ZJCX)居群的遗传多样性最低。各居群近交系数(Fis)均为正值, 平均为0.566 5, 居群存在杂合子缺失且以近交或自交为主。6个居群当前有效群体大小分析显示, 江苏溧阳(JSLY)居群无法获得Ne值, 可能其Ne值为无限大, 也可能由于个体数量较少导致, 其余5个居群的当前有效群体大小在3.8 (3.6, 4.0)- 9.4 (8.4, 10.4)之间。当前有效群体大小均值(5.6)极低, 可能在过去经历了瓶颈效应, 导致短穗竹分布区发生严重的居群收缩。
表2 6个居群遗传多样性指标
Table 2
| Population | PIC | Ho | He | π | Fis | Ne (95% CI) |
|---|---|---|---|---|---|---|
| AHYX | 0.8102 | 0.0880 | 0.3051 | 0.3295 | 0.6466 | 5.4 (5.2, 5.7) |
| AHHS | 0.6412 | 0.0863 | 0.2712 | 0.3099 | 0.4899 | 9.4 (8.4, 10.4) |
| AHGD | 0.8802 | 0.0885 | 0.3148 | 0.3339 | 0.7123 | 3.8 (3.6, 4.0) |
| JSLY | 0.5502 | 0.0841 | 0.2492 | 0.2990 | 0.4098 | Inf (inf, inf) |
| JSYX | 0.9069 | 0.0884 | 0.3183 | 0.3358 | 0.7385 | 4.0 (3.9, 4.2) |
| ZJCX | 0.5462 | 0.0865 | 0.2474 | 0.2969 | 0.4017 | 4.8 (4.5, 5.1) |
| Mean | 0.7225 | 0.0870 | 0.2843 | 0.3175 | 0.5665 | 5.6 (5.1, 5.9) |
AHYX、AHHS、AHGD、JSLY、JSYX和ZJCX同
AHYX, AHHS, AHGD, JSLY, JSYX, and ZJCX are the same as shown in
2.2 居群遗传结构
通过系统发育和遗传结构分析、主成分分析(PCA)和主成分判别分析(DAPC)明确了短穗竹现有居群遗传结构分布格局。系统发育树包括2个主要分支(图1A), 安徽霍山(AHHS)和安徽岳西(AHYX)两居群地理位置相对较近, 最先分离, 表明两居群之间亲缘关系较近。安徽广德(AHGD)、江苏溧阳(JSLY)、江苏宜兴(JSYX)和浙江长兴(ZJCX) 4个居群聚为一支, 它们可能拥有共同的祖先, 但个体间存在交叉混合。
图1
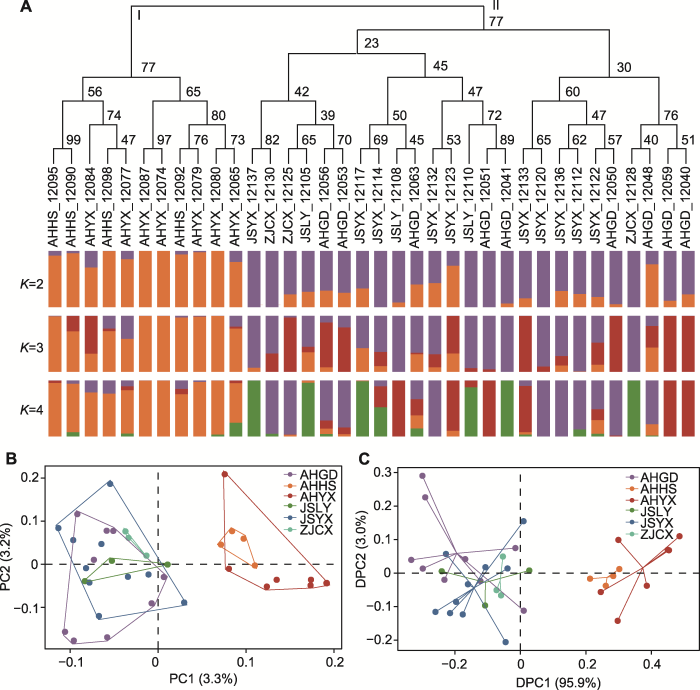
图1
短穗竹居群遗传结构分析
(A) 系统进化树和K=2, 3, 4时每个个体的遗传成分, 分支上方的数字表示靴带值; (B) 主成分分析(PCA); (C) 主成分判别分析(DAPC)。AHYX、AHHS、AHGD、JSLY、JSYX和ZJCX同
Figure 1
Population genetic structure analysis of Brachystachyum densiflorum
(A) Phylogenetic tree and individual genetic components at K=2, 3, 4, the numbers above the branch are the bootstrap values; (B) Principal component analysis (PCA); (C) Discriminant analysis of principal components (DAPC). AHYX, AHHS, AHGD, JSLY, JSYX, and ZJCX are the same as shown in
遗传结构分析表明, 遗传聚类值K=2时交叉检验错误率(CV error)最低(图2), 说明遗传物质来源于2个祖先并表现出稳定的遗传结构(图1A)。随着K值(3-4)的增加, 有不同的遗传信息加入, 并在安徽广德(AHGD)、江苏溧阳(JSLY)、江苏宜兴(JSYX)和浙江长兴(ZJCX) 4个居群中表现最明显, 安徽霍山(AHHS)和安徽岳西(AHYX)两居群遗传结构较稳定(图1A)。主成分分析(PCA)显示, 前2个轴分别解释3.3%和3.2%的遗传变异, 安徽霍山(AHHS)和安徽岳西(AHYX) 2个居群与其余4个居群分别形成独立的聚类且二者之间存在明显的遗传分化(图1B)。利用上述3种分析方法所得结果一致, 主成分判别分析(DAPC)进一步验证了短穗竹当前遗传结构的稳定性, 并确定短穗竹2个组的划分(图1C)。
图2
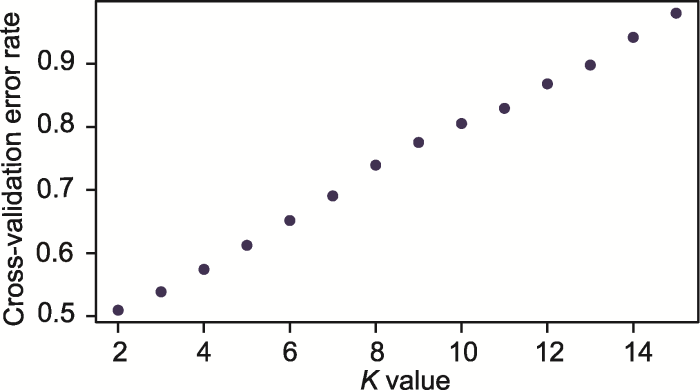
短穗竹居群存在中度遗传分化(FST=0.102, 0.05< FST<0.15), 遗传分化值(FST)介于0.048-0.151之间(表3)。江苏溧阳(JSLY)和浙江长兴(ZJCX)两居群虽然地理位置相近, 但居群间的遗传分化值(FST=0.151)是所有群体中最大的。分子方差分析(AMOVA)显示(附表5), 遗传变异主要发生在居群内(Va=79.71%), 居群间变异很小(Va=0.45%)。尽管各居群存在中度的遗传分化(FST=0.102), 但是基因流水平很高(Nm=2.442, Nm>1), 说明居群间存在一定的遗传交换, 从而抵消了基因漂变造成的强烈遗传分化(表3)。但是, 短穗竹各居群间近期(1-3代)基因流动率较低(附表5), 其范围在0.020 7-0.037 4之间, 平均值为0.029 4。
表3 6个居群间遗传分化值(FST, 左下三角)和基因流(Nm, 右上三角)
Table 3
| Population | AHGD | AHYX | AHHS | JSLY | JSYX | ZJCX |
|---|---|---|---|---|---|---|
| AHGD | 0.000 | 3.596 | 2.497 | 2.065 | 4.958 | 2.108 |
| AHYX | 0.065 | 0.000 | 2.382 | 1.869 | 3.848 | 1.887 |
| AHHS | 0.091 | 0.095 | 0.000 | 1.523 | 2.591 | 1.562 |
| JSLY | 0.108 | 0.118 | 0.141 | 0.000 | 2.154 | 1.406 |
| JSYX | 0.048 | 0.061 | 0.088 | 0.104 | 0.000 | 2.177 |
| ZJCX | 0.106 | 0.117 | 0.138 | 0.151 | 0.103 | 0.000 |
AHYX、AHHS、AHGD、JSLY、JSYX和ZJCX同
AHYX, AHHS, AHGD, JSLY, JSYX, and ZJCX are the same as shown in
2.3 遗传变异与气候关联分析
图3
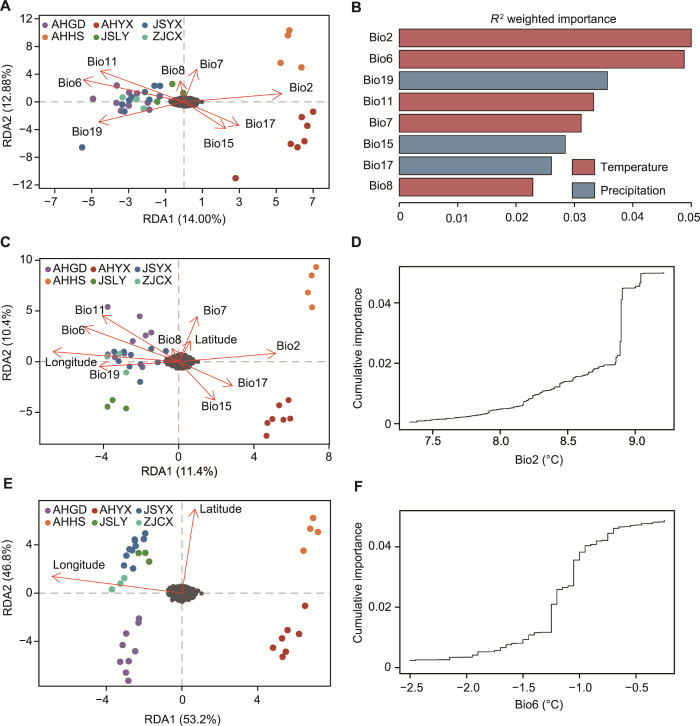
图3
冗余分析(RDA)和梯度森林(GF)分析
(A), (C), (E) 气候和地理与遗传结构之间的关联分析; (B) 梯度森林分析; (D), (F) Bio2和Bio6响应曲线。Bio2: 平均气温日较差; Bio6: 最冷月份最低温度; Bio7: 气温年较差; Bio8: 最湿季度平均温度; Bio11: 最冷季度平均温度; Bio15: 降水量季节性变化; Bio17: 最湿季度降水量; Bio19: 最冷季度降水量。AHYX、AHHS、AHGD、JSLY、JSYX和ZJCX同
Figure 3
Redundancy analysis (RDA) and gradient forest (GF) analysis
(A), (C), (E) Association analysis between climate, geography and genetic structure; (B) Gradient forest analysis; (D), (F) Bio2 and Bio6 response curves. Bio2: Average daily temperature range; Bio6: The lowest temperature in the coldest month; Bio7: Annual temperature range; Bio8: The average temperature of the wettest quarter; Bio11: The average temperature of the coldest quarter; Bio15: Seasonal variation of precipitation; Bio17: The wettest season precipitation; Bio19: The coldest season precipitation. AHYX, AHHS, AHGD, JSLY, JSYX, and ZJCX are the same as shown in
基于冗余分析和梯度森林分析, 将8个气候因子与短穗竹的遗传信息联系起来, 进一步确定影响其居群间遗传变异的因子(图3A, B)。冗余分析表明, RDA1和RDA2轴分别解释14.00%和12.88%的遗传变异(图3A)。RDA1轴中Bio6和Bio2的载荷最高(图3A), RDA2轴主要受到Bio11和Bio7的影响(附表6), 表明遗传变异与温差和低温之间密切相关。利用ordistep函数逐步排除法筛选可解释遗传变异的独立因子(附表6), 表明Bio19也是显著的预测因子(P< 0.05, Radj2=0.004 4), 这有助于解释遗传结构的差异。梯度森林分析表明, Bio2是最重要的预测因子, 其次是Bio6 (图3B)。Bio2在8.2-9.0°C时发生变化, 在8.9°C时突然变化(图3D); Bio6在-2.1- -0.7°C时发生变化, -2.1- -1.4°C变化较为平缓, -1.2- -0.7°C变化幅度较大(图3F)。此外, 排在前5位的气候因子中还有最冷季度降水量(Bio19), 说明降水和低温影响短穗竹的遗传变异。
综合冗余分析和梯度森林分析, 表明遗传变异与5个气候因子(Bio2、Bio6、Bio19、Bio11和Bio7)之间存在显著关联, 共同驱动短穗竹的遗传变异。本研究筛选出短穗竹居群适应性位点(异常SNPs) 617个, 去除重复后剩余544个。利用BayeScan软件基于贝叶斯概率法检测出98个潜在的候选位点(图4A)。通过冗余分析识别出170个异常SNPs (图4B), 其中57个适应Bio19, 37个适应Bio2, 26个适应Bio6, 26个适应Bio7, 24个适应Bio11。基于潜在因素混合模型(LFMM) 筛选出349个异常SNPs (图4C), 其中217个与Bio19相关, 99个与Bio7相关, 23个与Bio11相关, 6个与Bio2相关, 4个与Bio6相关。冗余分析和偏冗余分析(pRDA)进一步验证了适应性位点与5个气候因子之间的关联性(附表7), 结果显示适应性位点与温差、低温(Bio2、Bio6、Bio11和Bio7)和最冷季度降水量呈(Bio19)极显著相关(Radj2=0.034, P<0.01)。
图4
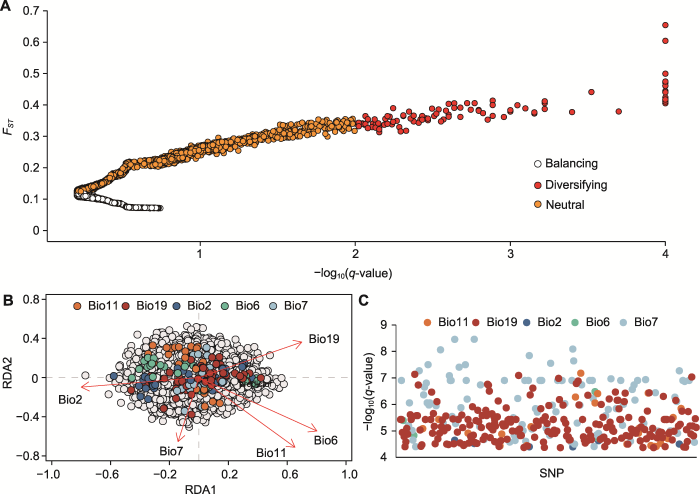
图4
利用3种方法筛选的异常单核苷酸多态性(SNP)位点
(A) BayeScan软件筛选; (B) 冗余分析(RDA)筛选; (C) 潜在因素混合模型(LFMM)筛选。FST: 遗传分化指数。Bio2、Bio6、Bio7、Bio11和Bio19同
Figure 4
Screening of outlier single nucleotide polymorphism (SNP) sites by three methods
(A) Screening by BayeScan software; (B) Screening by redundancy analysis (RDA); (C) Screening by latent factor mixed modeling (LFMM). FST: Fixation index of subdivision. Bio2, Bio6, Bio7, Bio11, and Bio19 are the same as shown in
2.4 潜在地理分布预测
MaxEnt模型能够准确预测短穗竹不同时期的适生区域(AUC值>0.9), 其ROC曲线下方面积AUC值为0.976, 平均标准偏差为0.006 (附图2)。基于综合贡献率、置换重要值(附表8)和刀切法检验(附图3), 结果表明短穗竹的分布和生长主要受Bio6、Bio11、Bio17和Bio19四个因子的影响。
通过MaxEnt模型预测短穗竹在末次盛冰期、全新世中期、当前和未来的潜在分布区, 发现从末次盛冰期(图5C)、全新世中期(图5B)到当前(图5A)短穗竹栖息地随着时间的推移发生了巨大变化, 适生区波动明显, 其分布范围不断扩大, 由1.06×105 km2增大到1.02×106 km2, 分布面积增加了89.5%且明显向北迁移(附表9; 图5A-C),说明短穗竹在末次盛冰期和全新世中期不同气候条件下通过迁移持续进行适应性生长并延续至今。当前气候条件下, 短穗竹实际分布点被适生区覆盖, 且集中分布于高适生区内(图5A); 适生区分布范围在108°-120°E、23°-34°N之间, 其中高适生区面积为1.9×105 km2 (附表9), 主要位于浙江、安徽中部至南部和江苏南部, 江西和湖南交界处也有分布(图5A)。未来气候条件下, 在2021-2040年(图5D, G)和2041-2060年(图5E,H) 2个时段, 相比当前适生区面积和分布区波动将较小。在SSP 1-2.6路径下高适生区有向北迁移的趋势(图5D, E), 在2021- 2040年高适生区面积将略有增加, 但在2041-2060年将减小。预计2061-2080年(图5F, I), 高适生区范围将缩小, 特别是在SSP 5-8.5路径下, 短穗竹在湖南的适生区将明显收缩, 在安徽高适生区将出现部分衰退和破碎化(图5I)。
图5
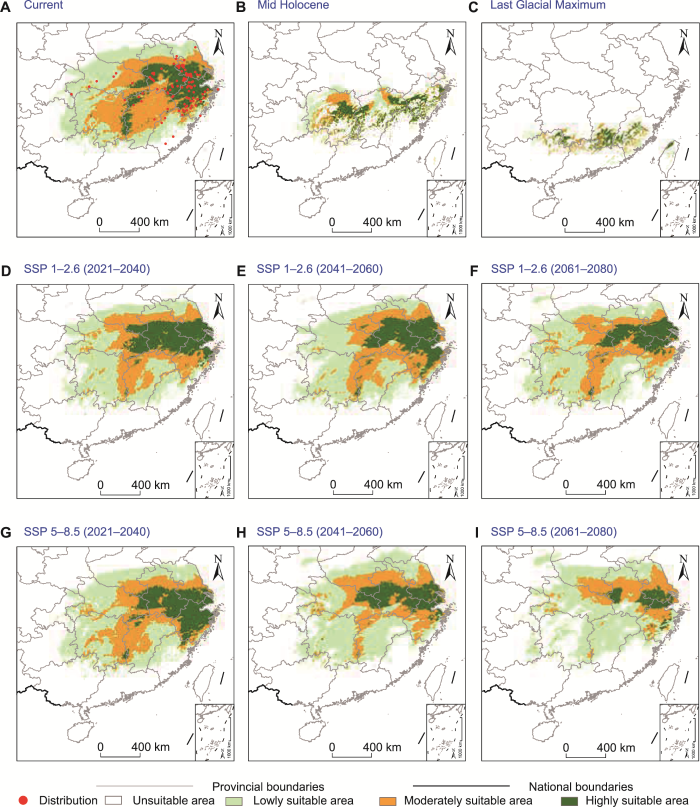
图5
不同时期短穗竹适宜性生境分布
(A) 当前(1970-2000年)气候条件下; (B) 全新世中期气候条件下; (C) 末次盛冰期气候条件下; (D), (G) 2021-2040年间气候; (E), (H) 2041-2060年间气候; (F), (I) 2061-2080年间气候
Figure 5
Distribution of suitable habitats for Brachystachyum densiflorum in different periods
(A) Current (1970‒2000) climate scenarios; (B) Mid Holocene climate scenarios; (C) Last Glacial Maximum climate scenarios; (D), (G) 2021-2040 climate scenarios; (E), (H) 2041-2060 climate scenarios; (F), (I) 2061-2080 climate scenarios
3 讨论
3.1 短穗竹遗传多样性
遗传多样性水平反映了物种短期生态适应和长期进化所需遗传资源的可用性(Meena et al., 2019)。竹类的遗传多样性表现在广泛的变异(Perez-Alquicira et al., 2021)。本研究中, 短穗竹具有中等水平的遗传多样性(PIC=0.722 5, Ho=0.087, He=0.284 3, π=0.317 5), 遗传多样性水平高于RAD测序的2种热带木本竹类澜沧梨藤竹(Ho=0.160, He=0.162, π=0.174)和独龙江空竹(Ho=0.217, He=0.252, π=0.257) (赵虎刚等, 2023; 张如礼等, 2024)。多种因素决定了物种的遗传多样性, 多倍体是使竹类保持高水平遗传多样性的因素之一, 原因是每个基因座的等位基因数量较多(Perez-Alquicira et al., 2021)。目前, 已在基因组水平证实木本竹类是不均匀的多倍体, 短穗竹隶属青篱竹族(Arundinarieae), 是四倍体温带木本竹类(2n=46- 48)的一员(Guo et al., 2019; Clark, 2023)。地理分布范围广或多年生的物种保持较高的遗传多样性, 短穗竹在江苏、浙江、安徽、福建、湖北和江西均有分布(附表3)。木本竹类也是禾本科乃至单子叶植物中寿命最长的物种, 其寿命长达15-150年, 广泛而连续的分布和较长的寿命使得短穗竹具有丰富的遗传多样性(Meena et al., 2019; Oumer et al., 2020; Poupon et al., 2021; Clark, 2023)。此外, 克隆植物居群可能具有多个多源基因型, 并保持较高的遗传多样性。竹类繁殖多以克隆为主且进化速率较慢, 基因组变异较小, 现有居群可能保留了其祖先的高遗传多样性, 尽管最近的气候变化和人为压力导致居群数量减少, 但居群仍表现出较高的遗传多样性(Yang et al., 2012; Das et al., 2017; Jiang et al., 2017; Li et al., 2020; Clark, 2023 )。取样不足或取样范围较小也会影响对遗传多样性的评估。本研究中短穗竹个体主要采集于当前的分布中心区域(表1), 且每个居群个体较少(3-10个)。地理代表性取样不够全面可能会影响等位基因的获取, 导致评价结果无法反映出居群整体的遗传多样性水平(Wang et al., 2017; 张如礼等, 2024)。
3.2 居群遗传结构和遗传分化
本研究中, 通过遗传结构分析将6个居群划分为2组, 安徽霍山(AHHS)和安徽岳西(AHYX) 2个居群与其余4个居群之间有很强的遗传结构分化(图1)。基因流受限是居群遗传结构形成的主要原因, 而地理距离较远阻碍了2个组之间的连通性, 如安徽霍山(AHHS)和安徽岳西(AHYX) 2个居群与其余4个居群地理距离较远(274.4-346.42 km), 阻碍了2个组之间的基因交流(Hardy et al., 2006; Perez-Alquicira et al., 2021; Wang et al., 2022a)。此外, 居群的历史事件、自然选择和人为压力也会促进居群遗传结构的形成(Cheng et al., 2020)。研究还发现地理分布区域邻近的居群遗传背景具有较高的相似性, 大多数竹类居群可通过无性繁殖建立并扩展到附近区域, 共享相同的基因库, 因此居群间的亲缘关系较近, 同源性较高(Meena et al., 2019; Oumer et al., 2020; Silva et al., 2020)。本研究中短穗竹取样个体并未完全覆盖分布区域内的所有居群(附表3), 除当前的分布中心(表1)区域外, 尚缺乏对福建、江西、湖北、江苏北部及浙江南部等地的取样(图5A), 可能会造成对居群遗传结构评价的偏差, 从而无法准确判断居群间的遗传关系。因此, 增加地理代表性分布区域内居群取样并确保每个居群有足够的个体数量, 从而增加遗传信息数量, 提高物种居群遗传结构预测的准确性(Perez-Alquicira et al., 2021)。
短穗竹居群较大的遗传变异比例局限在居群内(79.71%), 但依然存在中度遗传分化(FST=0.102), 在一些竹类中也存在中度遗传分化, 如须弥筱竹(Himalayacalamus falconeri) (FST=0.121)、瓜多竹(FST= 0.098)和Kuruna debilis (FST=0.113) (Attigala et al., 2017; Perez-Alquicira et al., 2021; Meena et al., 2023b)。居群间的遗传分化与基因流动呈负相关, 尽管短穗竹居群之间基因流水平很高(Nm=2.442), 但地理分布较近的江苏溧阳(JSLY)与浙江长兴(ZJCX)的居群存在较大分化(FST=0.151>0.15)。即使地理位置接近的居群, 由于开花时间不同步、开花不频繁及花的数量少等原因造成花粉传播率低, 可能会阻碍基因流动, 促进居群间的分化(Nilkanta et al., 2017; Perez-Alquicira et al., 2021)。人为压力增大及全球气候变化等原因导致栖息地退化和破碎化可能加剧江苏溧阳(JSLY)和浙江长兴(ZJCX)两居群的遗传漂变, 从而导致基因交流减弱(近期基因流动率为0.029 4), 因而表现出更强的遗传分化(Nilkanta et al., 2017; Meena et al., 2019)。
3.3 遗传变异与气候关联分析
冗余分析和梯度森林分析表明, 短穗竹适应性遗传变异很大程度上与Bio2、Bio6、Bio7、Bio11和Bio19这5个气候因子紧密关联, 温差和低温是影响短穗竹遗传变异的关键因子, 此外也与最冷季度降水量(Bio19)有关。温度是许多物种存在适应性变化的驱动因素, 其通过影响光合作用、呼吸作用和蒸腾作用影响植物的生长(Jia et al., 2020; Butler et al., 2022)。温度和降水是影响植物生长发育、生存和繁殖的重要因素, 但还需综合考虑其它相关因素, 如土壤特性和光照辐射, 这可能会改变对适应性遗传变异的认识和理解(Perez-Alquicira et al., 2021; Filipe et al., 2022)。本研究基于16 583个SNPs筛选适应性位点(异常SNP), 但由于大多数SNP是中性的, 仅发现98个高分化的SNP位点(图4A)。 Wang等(2017)认为利用简化基因组测序技术在基因组水平上筛选适应性位点有一定的局限性, 可能无法发现与适应性有关的低频等位基因。通过冗余分析和潜在因素混合模型筛选, 发现与降水量(Bio19)有关的异常SNP比各温度因子多(图4B, C)。气候因子可能不是某些假定的适应性SNP变异的主要驱动因素, 相关SNP可能具有多效性并受到其它生物或非生物因素的选择(Filipe et al., 2022)。
3.4 潜在地理分布预测
第四纪气候在冰期和间冰期之间振荡对当今物种的分布格局产生重要影响。许多植物无法适应寒冷干燥的气候而被迫向南迁移避难, 众多物种被隔离在不同的避难所(胡菀等, 2020; Polic et al., 2022)。在末次冰期许多植物并未完全迁移到24°N以南区域, 在末次盛冰期大降温背景下短穗竹的分布范围在23°- 27°N之间, 说明该分布区(图5C)可能是短穗竹在冰期的避难所(胡菀等, 2020)。全新世中期气候开始变暖, 一些物种向北迁移并且分布面积增大(胡菀等, 2020; He et al., 2022)。短穗竹在全新世中期明显向北扩张且分布范围扩大59.12% (图5B; 附表9)。全新世中期到当前短穗竹同样向北迁移和扩张(图5A, B), 此期间温度降低可能是影响短穗竹扩张的原因, 已证实全新世中期(约6 000年前)到当前发生了5次冷事件, 分别发生在5.9、4.2、2.8、1.4和0.4 kyr (Wang et al., 2013)。在进行短穗竹分布预测时也发现最冷月份最低温度(Bio6)、最冷季度平均温度(Bio11)和最冷季度降水量(Bio19)影响短穗竹的分布和生长。未来气候条件下, 物种的承受能力将受降水和温度等因素的影响, 物种的分布范围可能会缩小或转移(Yebeyen et al., 2022)。2021-2040年和2041-2060年2个时段, 在SSP 1-2.6路径下短穗竹高适生区有向北迁移的趋势。此外, 预测江西和湖南交界处的高适生区面积将减小甚至消失, 以及在2061-2080年间SSP 5-8.5路径下高适生区会出现部分衰退和破碎化, 短穗竹可能将受到全球变暖的严重威胁。
作者贡献声明
张如礼: 撰写论文, 完成数据分析和图表绘制; 李德铢: 构思主题和设计论文框架, 修改并完善论文; 张玉霄: 样品采集, 构思主题和设计论文框架, 修改并完善论文, 定稿。
附表1 从短穗竹采样地点经纬度提取的气候数据
Appendix table 1 Climate data extracted from latitude and longitude of the sampled sites of Brachystachyum densiflorum
附表2 19个气候因子相关性分析
Appendix table 2 Correlation analysis of 19 climate factors
附表3 短穗竹分布位置筛选
Appendix table 3 Screening of the distribution of Brachystachyum densiflorum
附表4 要素类型与正则化乘数
Appendix table 4 The feature combinations and the regularization multipliers
附表5 分子方差分析
Appendix table 5 Analysis of molecular variance (AMOVA)
附表6 地理和气候、气候、地理3组数据对短穗竹遗传结构影响对比
Appendix table 6 Comparison of the effects on the genetic structure of Brachystachyum densiflorum of three data sets: climate and geography, climate, geography
附表7 适应性位点冗余分析和偏冗余分析
Appendix table 7 Redundancy analysis and partial redundancy analysis of adaptive loci
附表8 8个气候因子及其重要性参数
Appendix table 8 Eight climate factors and their importance parameters
附表9 不同时期短穗竹的潜在分布面积
Appendix table 9 The potential distribution area of Brachystachyum densiflorum under different periods
附图1 SNP转换与颠换统计
Appendix figure 1 Transition and transversion statistics of SNP
附图2 MaxEnt模型中的ROC预测
Appendix figure 2 Prediction of the ROC in the MaxEnt model
附图3 气候变量对短穗竹潜在分布模型的Jackknife检验得分
Appendix figure 3 Jackknife test scores of climate variables on potential distribution model of Brachystachyum densiflorum

参考文献

Climate change is a threat to biodiversity. One way that this threat manifests is through pronounced shifts in the geographical range of species over time. To predict these shifts, researchers have primarily used species distribution models. However, these models are based on assumptions of niche conservatism and do not consider evolutionary processes, potentially limiting their accuracy and value. To incorporate evolution into the prediction of species' responses to climate change, researchers have turned to landscape genomic data and examined information about local genetic adaptation using climate models. Although this is an important advancement, this approach currently does not include other evolutionary processes-such as gene flow, population dispersal and genomic load-that are critical for predicting the fate of species across the landscape. Here, we briefly review the current practices for the use of species distribution models and for incorporating local adaptation. We next discuss the rationale and theory for considering additional processes, reviewing how they can be incorporated into studies of species' responses to climate change. We summarize with a conceptual framework of how manifold layers of information can be combined to predict the potential response of specific populations to climate change. We illustrate all of the topics using an exemplar dataset and provide the source code as potential tutorials. This Perspective is intended to be a step towards a more comprehensive integration of population genomics with climate change science.© 2021. Springer Nature Limited.
Identifying areas of high evolutionary potential is a judicious strategy for developing conservation priorities in the face of environmental change. For wide-ranging species occupying heterogeneous environments, the evolutionary forces that shape distinct populations can vary spatially. Here, we investigate patterns of genomic variation and genotype-environment associations in the hermit thrush (), a North American songbird, at broad (across the breeding range) and narrow spatial scales (at a hybrid zone). We begin by building a genoscape or map of genetic variation across the breeding range and find five distinct genetic clusters within the species, with the greatest variation occurring in the western portion of the range. Genotype-environment association analyses indicate higher allelic turnover in the west than in the east, with measures of temperature surfacing as key predictors of putative adaptive genomic variation rangewide. Since broad patterns detected across a species' range represent the aggregate of many locally adapted populations, we investigate whether our broadscale analysis is consistent with a finer scale analysis. We find that top rangewide temperature-associated loci vary in their clinal patterns (e.g., steep clines vs. fixed allele frequencies) across a hybrid zone in British Columbia, suggesting that the environmental predictors and the associated candidate loci identified in the rangewide analysis are of variable importance in this particular region. However, two candidate loci exhibit strong concordance with the temperature gradient in British Columbia, suggesting a potential role for temperature-related barriers to gene flow and/or temperature-driven ecological selection in maintaining putative local adaptation. This study demonstrates how patterns identified at the broad (macrogeographic) scale can be validated by investigating genotype-environment correlations at the local (microgeographic) scale. Furthermore, our results highlight the importance of considering the spatial distribution of putative adaptive variation when assessing population-level sensitivity to climate change and other stressors.© 2022 The Authors. Evolutionary Applications published by John Wiley & Sons Ltd.
In conservation biology genetic diversity is recognized as an important criterion to consider when prioritizing populations for protection. Today, population genomics offers the opportunity to evaluate both neutral and adaptive components of genetic diversity directly at the genome level with molecular tools. By screening the genome with many genetic markers, it is possible to detect loci supposedly under natural selection and thus of adaptive significance. We devised a new diversity index, the population adaptive index (PAI), which accounts for the adaptive value of the population it refers to. To estimate this index, we performed a genome scan with amplified fragment length polymorphism markers to identify neutral and selected loci in several populations of a widespread amphibian (common frog, Rana temporaria) and a threatened plant (Austrian dragonhead, Dracocephalum austriacum L.). We then investigated four different conservation strategies aimed at protecting the maximum amount of genetic diversity (neutral or selected). In particular we explored the relevance of the principle of complementarity, usually applied to the protection of species, in the management of intraspecific diversity. This principle advocates the conservation of sets of units that together maximize the species' or genetic diversity, which is in opposition to the traditional approach of targeting populations that are the most diverse individually. Four major conclusions emerged from these results. First, the PAI seemed to be a valuable index to evaluate the adaptive diversities within populations. Second, in the two species, the neutral and adaptive diversities within and among populations were not correlated, so conservation strategies based on the neutral and adaptive indexes would not select the same populations for protection. Third, because of its efficiency in conserving genetic diversity, the principle of complementarity deserves to be used more often for this purpose. Fourth, when neutral and adaptive results conflict, additional arguments (e.g., demography, ecology, and geographic proximity) should be considered together with levels of genetic diversity to determine a conservation strategy.
Studying population genetic structure and gene flow of plant populations and their influencing factors is of particular significance in the field of conservation biology, especially important for species such as rare and endangered plants. Tetraena mongolica Maxim. (TM), belongs to Zygophyllaceae family, a rare and endangered plant with narrow distribution. However, for the last decade, due to excessive logging, urban expansion, industrial and tourism development, habitat fragmentation and loss of natural habitats have become major threats to the population of endangered plants.In this study, genetic diversity, population genetic structure and gene flow of TM populations were evaluated by reduced representation sequencing technology, and a total of more than 133.45 GB high-quality clean reads and 38,097 high-quality SNPs were generated. Analysis based on multiple methods, we found that the existing TM populations have moderate levels of genetic diversity, and very low genetic differentiation as well as high levels of gene flow between populations. Population structure and principal coordinates analysis showed that 8 TM populations can be divided into two groups. The Mantel test detected no significant correlation between geographical distances and genetic distance for the whole sampling. Moreover, the migration model indicated that the gene flow is more of a north to south migration pattern in history.This study demonstrates that the present genetic structure is mainly due to habitat fragmentation caused by urban sprawl, industrial development and coal mining. Our recommendation with respect to conservation management is that, all 8 populations should be preserved as a whole population, rather than just those in the core area of TM nature reserve. In particular, the populations near the edge of TM distribution in cities and industrial areas deserve our special protection.
The advent of next-generation sequencing (NGS) has revolutionized genomic and transcriptomic approaches to biology. These new sequencing tools are also valuable for the discovery, validation and assessment of genetic markers in populations. Here we review and discuss best practices for several NGS methods for genome-wide genetic marker development and genotyping that use restriction enzyme digestion of target genomes to reduce the complexity of the target. These new methods -- which include reduced-representation sequencing using reduced-representation libraries (RRLs) or complexity reduction of polymorphic sequences (CRoPS), restriction-site-associated DNA sequencing (RAD-seq) and low coverage genotyping -- are applicable to both model organisms with high-quality reference genome sequences and, excitingly, to non-model species with no existing genomic data.
Temperature and precipitation regimes are rapidly changing, resulting in forest dieback and extinction events, particularly in Mediterranean-type climates (MTC). Forest management that enhance forests' resilience is urgently required, however adaptation to climates in heterogeneous landscapes with multiple selection pressures is complex. For widespread trees in MTC we hypothesized that: patterns of local adaptation are associated with climate; precipitation is a stronger factor of adaptation than temperature; functionally related genes show similar signatures of adaptation; and adaptive variants are independently sorting across the landscape. We sampled 28 populations across the geographic distribution of Eucalyptus marginata (jarrah), in South-west Western Australia, and obtained 13,534 independent single nucleotide polymorphic (SNP) markers across the genome. Three genotype-association analyses that employ different ways of correcting population structure were used to identify putatively adapted SNPs associated with independent climate variables. While overall levels of population differentiation were low (F = 0.04), environmental association analyses found a total of 2336 unique SNPs associated with temperature and precipitation variables, with 1440 SNPs annotated to genic regions. Considerable allelic turnover was identified for SNPs associated with temperature seasonality and mean precipitation of the warmest quarter, suggesting that both temperature and precipitation are important factors in adaptation. SNPs with similar gene functions had analogous allelic turnover along climate gradients, while SNPs among temperature and precipitation variables had uncorrelated patterns of adaptation. These contrasting patterns provide evidence that there may be standing genomic variation adapted to current climate gradients, providing the basis for adaptive management strategies to bolster forest resilience in the future.© 2022 The Authors. Molecular Ecology published by John Wiley & Sons Ltd.
The wild currant tomato Solanum pimpinellifolium inhabits a wide range of abiotic habitats across its native range of Ecuador and Peru. Although it has served as a key genetic resource for the improvement of domestic cultivars, little is known about the genetic basis of traits underlying local adaptation in this species, nor what abiotic variables are most important for driving differentiation. Here we use redundancy analysis (RDA) and other multivariate statistical methods (structural equation modelling [SEM] and generalized dissimilarity modelling [GDM]) to quantify the relationship of genomic variation (6,830 single nucleotide polymorphisms [SNPs]) with climate and geography, among 140 wild accessions. RDA, SEM and GDM each identified environment as explaining more genomic variation than geography, suggesting that local adaptation to heterogeneous abiotic habitats may be an important source of genetic diversity in this species. Environmental factors describing temporal variation in precipitation and evaporative demand explained the most SNP variation among accessions, indicating that these forces may represent key selective agents. Lastly, by studying how SNP-environment associations vary throughout the genome (44,064 SNPs), we mapped the location and investigated the functions of loci putatively contributing to climatic adaptations. Together, our findings indicate an important role for selection imposed by the abiotic environment in driving genomic differentiation between populations.© 2020 John Wiley & Sons Ltd.
The extent of gene dispersal is a fundamental factor of the population and evolutionary dynamics of tropical tree species, but directly monitoring seed and pollen movement is a difficult task. However, indirect estimates of historical gene dispersal can be obtained from the fine-scale spatial genetic structure of populations at drift-dispersal equilibrium. Using an approach that is based on the slope of the regression of pairwise kinship coefficients on spatial distance and estimates of the effective population density, we compare indirect gene dispersal estimates of sympatric populations of 10 tropical tree species. We re-analysed 26 data sets consisting of mapped allozyme, SSR (simple sequence repeat), RAPD (random amplified polymorphic DNA) or AFLP (amplified fragment length polymorphism) genotypes from two rainforest sites in French Guiana. Gene dispersal estimates were obtained for at least one marker in each species, although the estimation procedure failed under insufficient marker polymorphism, limited sample size, or inappropriate sampling area. Estimates generally suffered low precision and were affected by assumptions regarding the effective population density. Averaging estimates over data sets, the extent of gene dispersal ranged from 150 m to 1200 m according to species. Smaller gene dispersal estimates were obtained in species with heavy diaspores, which are presumably not well dispersed, and in populations with high local adult density. We suggest that limited seed dispersal could indirectly limit effective pollen dispersal by creating higher local tree densities, thereby increasing the positive correlation between pollen and seed dispersal distances. We discuss the potential and limitations of our indirect estimation procedure and suggest guidelines for future studies.
Adaptive differences across species' ranges can have important implications for population persistence and conservation management decisions. Despite advances in genomic technologies, detecting adaptive variation in natural populations remains challenging. Key challenges in gene-environment association studies involve distinguishing the effects of drift from those of selection and identifying subtle signatures of polygenic adaptation. We used paired-end restriction site-associated DNA sequencing data (6,605 biallelic single nucleotide polymorphisms; SNPs) to examine population structure and test for signatures of adaptation across the geographic range of an iconic Australian endemic freshwater fish species, the Murray cod Maccullochella peelii. Two univariate gene-association methods identified 61 genomic regions associated with climate variation. We also tested for subtle signatures of polygenic adaptation using a multivariate method (redundancy analysis; RDA). The RDA analysis suggested that climate (temperature- and precipitation-related variables) and geography had similar magnitudes of effect in shaping the distribution of SNP genotypes across the sampled range of Murray cod. Although there was poor agreement among the candidate SNPs identified by the univariate methods, the top 5% of SNPs contributing to significant RDA axes included 67% of the SNPs identified by univariate methods. We discuss the potential implications of our findings for the management of Murray cod and other species generally, particularly in relation to informing conservation actions such as translocations to improve evolutionary resilience of natural populations. Our results highlight the value of using a combination of different approaches, including polygenic methods, when testing for signatures of adaptation in landscape genomic studies.© 2017 John Wiley & Sons Ltd.
Aims Tsoongiodendron odorum is an ancient relic species belonging to the family Magnoliaceae, but it is labelled endangered plant with extremely small populations and facing serious threats to its wild survival now. Using Ecological Niche Modelling (ENM) to hindcast historical changes in its distribution during the Last Glacial Maximum (LGM), this study aims to explore the impact of climate change on the distribution of T. odorum, and to evaluate the relationship between species distribution and environmental variables. The results of this study could contribute to the conservation of T. odorum in the context of global warming. Methods Based on 96 modern geographical distribution records and 8 bioclimatic variables, we simulated the potential distribution of T. odorum during the LGM, Mid-Holocene, present and future (period of 2061-2080 in the Representative Concentration Pathway 8.5 climate scenario) with MaxEnt model. The changes in species distribution through time were analyzed by SDM toolbox, while the importance of bioclimatic variables was evaluated by percent contribution, permutation importance and Jackknife test. Important findings (1) The highly suitable region for T. odorum was Nanling region, and this area might be the refuge where T. odorum survived in situ during the LGM because only slightly southward retreat in distribution was detected in this region during the LGM. (2) In the two warming climate scenarios (Mid-Holocene and future), the area of the suitable region was reduced, and the decrease of future distribution is greater than that during the Mid-Holocene, which suggests that warming climate might have a negative impact on the distribution of T. odorum. (3) Overall the stability of distribution range of T. odorum in each period indicates the climate adaptation of this species. Human activity or self-breeding problem was likely the significant cause leading to endangered condition. Guangdong and Guangxi should be regarded as priority conservation areas as shown by our results.
观光木(Tsoongiodendron odorum)是木兰科的古老残遗物种, 目前正面临严峻的生存威胁, 属于极小种群濒危植物。通过生态位模型(ENM)能够重建观光木地理分布格局的历史变迁, 探究气候变化对该物种分布的影响, 并了解其地理分布与气候需求间的关系, 从而为全球变暖背景下观光木的保护提供理论基础。该文基于96条现代分布记录和8个环境变量, 采用最大熵(MaxEnt)模型模拟观光木在末次盛冰期、全新世中期、现代和未来(2061-2080年, RCP 8.5)的潜在分布区, 利用SDM toolbox分析观光木的地理空间变化, 并综合贡献率、置换重要值和Jackknife检验来评估气候因子的重要性。研究结果表明: (1)观光木的高度适生区在南岭地区, 末次盛冰期时没有大尺度向南退缩, 很可能在山区避难所原地存活; (2)在全新世中期和未来两个增温的气候情境下, 观光木的分布区均表现为缩减, 其中未来分布的减幅更大, 表明气候变暖对观光木的生长有一定的负面影响; (3)总体上看, 观光木各个时期的地理分布范围相对稳定, 说明观光木对气候变化有一定的适应能力, 人为活动或自身繁育问题可能是致濒的重要原因, 并建议对广东和广西群体进行优先保护。
Over the past twenty years conservation genetics has progressed from being mainly a theory-based field of population biology to a full-grown empirical discipline. Technological developments in molecular genetics have led to extensive use of neutral molecular markers such as microsatellites in conservation biology. This has allowed assessment of the impact of genetic drift on genetic variation, of the level of inbreeding within populations, and of the amount of gene flow between or within populations. Recent developments in genomic techniques, including next generation sequencing, whole genome scans and gene-expression pattern analysis, have made it possible to step up from a limited number of neutral markers to genome-wide estimates of functional genetic variation. Here, we focus on how the transition of conservation genetics to conservation genomics leads to insights into the dynamics of selectively important variation and its interaction with environmental conditions, and into the mechanisms behind this interaction.Copyright 2010 Elsevier Ltd. All rights reserved.
Genetic diversity provides the basic substrate for evolution, yet few studies assess the impacts of global climate change (GCC) on intraspecific genetic variation. In this review, we highlight the importance of incorporating neutral and non-neutral genetic diversity when assessing the impacts of GCC, for example, in studies that aim to predict the future distribution and fate of a species or ecological community. Specifically, we address the following questions: Why study the effects of GCC on intraspecific genetic diversity? How does GCC affect genetic diversity? How is the effect of GCC on genetic diversity currently studied? Where is potential for future research? For each of these questions, we provide a general background and highlight case studies across the animal, plant and microbial kingdoms. We further discuss how cryptic diversity can affect GCC assessments, how genetic diversity can be integrated into studies that aim to predict species' responses on GCC and how conservation efforts related to GCC can incorporate and profit from inclusion of genetic diversity assessments. We argue that studying the fate of intraspecifc genetic diversity is an indispensable and logical venture if we are to fully understand the consequences of GCC on biodiversity on all levels.© 2012 Blackwell Publishing Ltd.
Background: Sporadic flowering contributes significantly to genetic diversity and connectivity among populations. Woody bamboos present sporadic or gregarious flowering patterns with long flowering cycles. In this study, we analyze the genetic diversity of three Guadua species distributed along the Gulf of Mexico slope that have different patterns of flowering.
Understanding which factors and processes are associated with genetic differentiation within and among species remains a major goal in evolutionary biology. To explore differences and similarities in genetic structure and its association with geographical and climatic factors in sympatric sister species, we conducted a large-scale (>32° latitude and >36° longitude) comparative phylogeographical study on three Argynnini butterfly species (Speyeria aglaja, Fabriciana adippe and F. niobe) that have similar life histories, but differ in ecological generalism and dispersal abilities. Analyses of nuclear (ddRAD-sequencing derived SNP markers) and mitochondrial (COI sequences) data revealed differences between species in genetic structure and how genetic differentiation was associated with climatic factors (temperature, solar radiation, precipitation, wind speed). Geographical proximity accounted for much of the variation in nuclear and mitochondrial structure and evolutionary relationships in F. adippe and F. niobe, but only explained the pattern observed in the nuclear data in S. aglaja, for which mitonuclear discordance was documented. In all species, Iberian and Balkan individuals formed genetic clusters, suggesting isolation in glacial refugia and limited postglacial expansion. Solar radiation and precipitation were associated with the genetic structure on a regional scale in all species, but the specific combinations of environmental and geographical factors linked to variation within species were unique, pointing to species-specific responses to common environments. Our findings show that the species share similar colonization histories, and that the same ecological factors, such as niche breadth and dispersal capacity, covary with genetic differentiation within these species to some extent, thereby highlighting the importance of comparative phylogeographical studies in sympatric sister species.© 2022 The Authors. Molecular Ecology published by John Wiley & Sons Ltd.
Rapid global climate change is posing a substantial threat to biodiversity. The assessment of population vulnerability and adaptive capacity under climate change is crucial for informing conservation and mitigation strategies. Here we generate a chromosome-scale genome assembly and re-sequence genomes of 230 individuals collected from 24 populations for Populus koreana, a pioneer and keystone tree species in temperate forests of East Asia. We integrate population genomics and environmental variables to reveal a set of climate-associated single-nucleotide polymorphisms, insertion/deletions and structural variations, especially numerous adaptive non-coding variants distributed across the genome. We incorporate these variants into an environmental modeling scheme to predict a highly spatiotemporal shift of this species in response to future climate change. We further identify the most vulnerable populations that need conservation priority and many candidate genes and variants that may be useful for forest tree breeding with special aims. Our findings highlight the importance of integrating genomic and environmental data to predict adaptive capacity of a key forest to rapid climate change in the future.© 2022. The Author(s).
The global loss of biodiversity continues at an alarming rate. Genomic approaches have been suggested as a promising tool for conservation practice as scaling up to genome-wide data can improve traditional conservation genetic inferences and provide qualitatively novel insights. However, the generation of genomic data and subsequent analyses and interpretations remain challenging and largely confined to academic research in ecology and evolution. This generates a gap between basic research and applicable solutions for conservation managers faced with multifaceted problems. Before the real-world conservation potential of genomic research can be realized, we suggest that current infrastructures need to be modified, methods must mature, analytical pipelines need to be developed, and successful case studies must be disseminated to practitioners. Copyright © 2014 Elsevier Ltd. All rights reserved.
理解物种的濒危机制对生物多样性的科学保护至关重要。荷叶铁线蕨(Adiantum nelumboides)作为国家一级重点保护野生植物, 其遗传多样性状况和濒危机制一直存在较大争议。本文利用简化基因组测序技术(genotyping by sequencing, GBS)对来自6个居群的28个荷叶铁线蕨样本测序, 共获得29.6 Gb的数据, 并筛选得到9,423个高质量单核苷酸变异位点(SNP), 通过遗传多样性和居群遗传结构分析, 并结合不同气候情景下物种潜在分布区差异, 探讨了荷叶铁线蕨的濒危原因和科学保护策略。结果表明: (1)荷叶铁线蕨具有较低的遗传多样性(H<sub>o </sub>= 0.138、H<sub>e </sub>= 0.232、P<sub>i </sub>= 0.373), 同时种群间具有较低的遗传分化(F<sub>st </sub>= 0.0202)和基因流(N<sub>m </sub>= 1.9613); (2)所有样本均来自2个遗传分组, 基因组大小为 5.01‒5.83 Gb, 且均为四倍体, GC含量约为 39%‒41%; (3)生态位模拟表明, 与现代气候相比, 在未来气候变化下荷叶铁线蕨的潜在分布区面积略有增加, 但高适生区面积减小。其主要适生区向北迁移, 影响其分布的主导因子为昼夜温差月均值和最冷季降水量。正是由于荷叶铁线蕨遗传多样性低, 不同种群间遗传分化较低, 再加上气候条件的变化, 其适生区狭窄, 导致其遗传多样性和种群数量急剧下降。因此, 自身更新能力低以及过度的人为活动干扰可能是导致其濒危的主要原因。建议加强对荷叶铁线蕨的就地保护; 通过生境恢复及自然回归等措施, 增加居群间的基因交流, 防止遗传资源丢失加剧。
极小种群野生植物云南蓝果树是国家和云南省实施极小种群野生植物保护工程的代表性物种。为有效保护其遗传资源,本研究通过二代测序技术,对其进行简化基因组测序,开发一批特异性高的单核苷酸多态性标记,分析现存群体的遗传结构和遗传多样性。经过遗传变异检测,本次研究中共获得SNP位点98 498个,通过样品最低测序深度>2,样品缺失率0.05筛选以后,得到有效SNP位点6 309个。基于过滤后的SNP,运用生物信息学分析方法,对云南蓝果树完成了群体的遗传分析,其中:系统进化树分析将云南蓝果树划分为3大类,研究分析了云南蓝果树各分类的私人等位基因数目(Private)、平均观测杂合度(H<sub>o</sub>)、平均期望杂合度(H<sub>e</sub>)、核苷酸多样性(π)和平均近交系数(F<sub>IS</sub>)5个遗传多样性参数;群体结构和主成分分析进一步证明了,云南蓝果树现存植株之间亲缘关系较远,遗传多样性差异较大,具有很高的遗传资源保存价值。本研究结果将为基于遗传管理的云南蓝果树就地保护、遗传资源保存和种群重建等保护工程提供科学依据。
Bamboo is an important member of the giant grass subfamily Bambusoideae of Poaceae. In this study, 13 bamboo accessions belonging to 5 different genera were subjected to morphological evaluation and sequence-related amplified polymorphism (SRAP) analysis. Unweighted pair-group method of arithmetic averages (UPGMA) cluster analysis was used to construct a dendrogram and to estimate the genetic distances among accessions. On the basis of morphological characteristics, the 13 accessions were distinctly classified into 2 major clusters; 3 varieties, PPYX, PGNK, and PLYY were grouped as cluster A, and 10 accessions were categorized under cluster B. Similarity coefficients ranging from 0.23 to 0.96 indicated abundant genetic variation among bamboo varieties. Approximately 38 SRAP primer combinations generated 186 bands, with 150 bands (80.65%) showing polymorphisms among the 13 accessions. Based on SRAP analysis, 13 bamboo accessions were grouped into 3 major clusters. Five species comprised Cluster I (PASL, PLYY, PTSC, SCNK, and BMAK), which belongs to genus Phyllostachys. Cluster II consisted of 5 varieties, PASL, PLYY, PTSC, SCNK, and BMAK; Cluster III included 3 varieties, PGNK, PLSY, and BMRS. Comparison of the results generated by morphological and SRAP analyses showed that the classification based on SRAP markers was more concordant to the taxonomic results of Gamble than that performed using morphological characters, thus suggesting that SRAP analysis is more efficient in evaluating genetic diversity in bamboos compared to morphological analysis. The SRAP technique serves as an alternative method in assessing genetic diversity within bamboo collections.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}