
 首页
首页

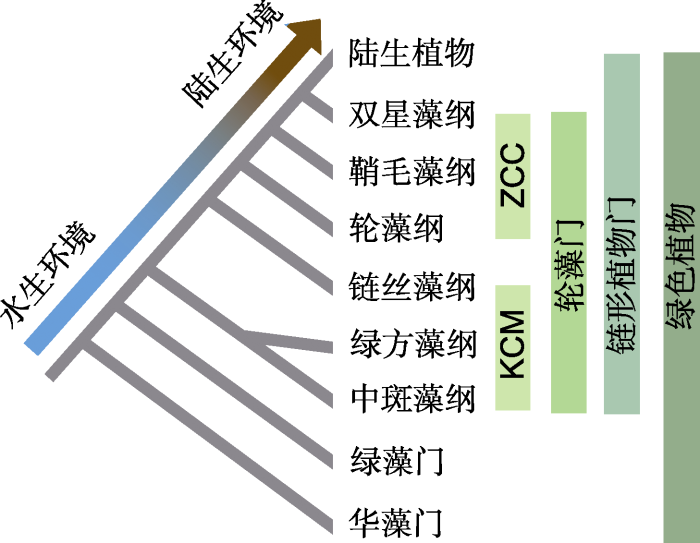
轮藻植物在15世纪60年代末被发现, 其分类归属一直存在争议。直到1879年, Migula建议将轮藻独立为轮藻门(Charophyta) (Greene, 1887)。轮藻门植物包含6个纲, 分别为单细胞或丝状的双星藻纲 (Zygnematophyceae)、有薄片组织的鞘毛藻纲(Coleochaetophyceae)、体型较大的轮藻纲(Charophyceae)、呈丝状的链丝藻纲(Klebsormidiophyceae)、可形成肉芽孢囊的绿方藻纲(Chlorokybophyceae)和单细胞的中斑藻纲(Mesostigmatophyceae) (Fürst-Jansen et al., 2020)。轮藻门植物处于绿色植物(Viridiplantae)进化树的关键节点(图1)。轮藻门植物与陆生植物(Embryophyta)共同组成单系群链形植物门(Streptophyta) (吴珍和程时锋, 2021; Domozych and Bagdan, 2022)。链形植物门与绿藻门(Chlorophyta)和华藻门(Prasinodermophyta)组成绿色植物(Li et al., 2020; Jiao et al., 2020)。现存的轮藻门植物可分为2个组: KCM组包括链丝藻纲、绿方藻纲和中斑藻纲, 是较早分化出的轮藻植物; ZCC组包括双星藻纲、鞘毛藻纲和轮藻纲, 是较晚分化出的轮藻植物(de Vries and Archibald, 2018)。现有的化石证据和分子证据表明, 所有现生的陆生植物是在5-6亿年前从单一的轮藻植物祖先演化而来(Fürst-Jansen et al., 2022)。因此对轮藻门植物的系统研究对于理解陆生植物的登陆过程至关重要。
图1
基因组是一个生物体内遗传物质的总和, 其蕴含着丰富完整的遗传信息(Hamilton and Buell, 2012)。植物基因组学研究在遗传信息解码的基础上, 为生态学、植物的起源与演化、农业生产、分子育种和关键基因挖掘等提供重要的基础信息(王英豪等, 2024)。随着高通量测序技术的快速发展, 以及基因组组装算法和生物信息学的进步, 在2000-2020年, 共有782种植物1 144个基因组进行了测序组装; 而在2021- 2023年, 共有1 031种植物2 373个基因组进行了测序组装, 其中包括793个新测序物种(Xie et al., 2024)。自从2014年第1个轮藻Klebsormidium nitens基因组被测序(Hori et al., 2014)以来, 多种轮藻门植物的全基因组已完成测序和组装。基于轮藻参考基因组信息, 研究人员在揭示绿色植物演化, 尤其是陆生植物起源机制方面取得了重大进展。
1 轮藻植物全基因组与植物陆地化
表1 已完成全基因组测序的轮藻门植物及分类层级
Table 1
| Species | Class | Order | Family |
|---|---|---|---|
| Zygnema circumcarinatum | Zygnematophyceae | Zygnematales | Zygnemataceae |
| Z. cf. cylindricum | Zygnematophyceae | Zygnematales | Zygnemataceae |
| Mesotaenium endlicherianum | Zygnematophyceae | Zygnematales | Mesotaeniaceae |
| Spirogloea muscicola | Zygnematophyceae | Spirogloeales | Spirogloeaceae |
| Closterium peracerosum-strigosum-littorale complex | Zygnematophyceae | Desmidiales | Closteriaceae |
| Penium margaritaceum | Zygnematophyceae | Desmidiales | Peniaceae |
| Chara braunii | Charophyceae | Charales | Characeae |
| Klebsormidium nitens | Klebsormidiophyceae | Klebsormidiales | Klebsormidiaceae |
| Chlorokybus atmophyticus | Chlorokybophyceae | Chlorokybales | Chlorokybaceae |
| Mesostigma viride | Mesostigmatophyceae | Mesostigmatales | Mesostigmataceae |
1.1 双星藻纲物种全基因组
陆地化(terrestrialization)是指陆生植物藻类祖先从水生环境到陆地环境的逐步演化, 最终在不同的陆地生境中生存繁殖并且长期适应下来的过程(吴珍和程时锋, 2021)。此过程通常被认为是陆生植物群落演化和多样化中的关键事件, 它深刻地改变了地球表面, 推动了陆地生态系统的发展, 为后来的动物登陆和陆地生物多样性的丰富奠定了基础(Cheng et al., 2019; 薛进庄等, 2022; Feng et al., 2024)。陆生植物在5-6亿年前起源于轮藻门植物(Delwiche and Cooper, 2015; Fürst-Jansen et al., 2022)。由于轮藻纲物种形态结构复杂, 并具有与根、茎、叶相似的分化结构以及特殊的有性生殖器官, 曾被认为是陆生植物的姐妹类群(Pringsheim, 1862)。基于对4个基因的系统发生分析, 认为轮藻纲Charales目是陆生植物的姐妹类群(Karol et al., 2001)。基于转录组和细胞器基因组的系统发生分析以及形态学研究, 目前认为双星藻纲是与现存陆生植物亲缘关系最近的姐妹类群(Wickett et al., 2014; Ruhfel et al., 2014; Puttick et al., 2018; One Thousand Plant Transcriptomes Initiative, 2019; Zhou and von Schwartzenberg, 2020)。因此, 双星藻纲物种的全基因组序列数据对于解析陆生植物的起源与演化具有重要价值。双星藻纲包括5个目(Hess et al., 2022), 其中已有4 426个物种被描述(Guiry, 2021; Guiry and Guiry, 2025)。目前有6个双星藻纲物种已完成全基因组测序, 其中Zygnema circumcarinatum的3个株系和Closterium peracerosum-strigosum-littorale complex的2个株系已测序, 其余物种仅1个株系完成测序(表2)。双星藻目(Zygnematales)和鼓藻目(Desmidiales)已测序物种均有2个, Spirogloeales目已测序物种有1个, 其余2个目(Serritaeniales和Spirogyrales)尚无已测序物种。
表2 双星藻纲轮藻植物基因组组装统计
Table 2
| Species (strain) | Predicted genome size (Mb) | Assembly size (Mb) | Number of scaffold | Scaffold N50 (kb) | GC content (%) | Number of gene | Reference |
|---|---|---|---|---|---|---|---|
| Zygnema circumcarinatum (SAG 698-1b) | 66.7 | 71.0 | 90 (20) | 3958.3 | 50.0 | 16617 | Feng et al., 2024 |
| Z. circumcarinatum (UTEX 1559) | 66.3 | 71.3 | 614 | 3970.3 | 49.5 | 18062 | Feng et al., 2024 |
| Z. circumcarinatum (UTEX 1560) | 67.8 | 67.3 | 514 | 3792.7 | 49.5 | 18654 | Feng et al., 2024 |
| Z. cf. cylindricum (SAG 698-1a_XF) | 322.5 | 359.8 | 3587 | 213.9 | 40.0 | 45178 | Feng et al., 2024 |
| Mesotaenium endlicherianum (SAG 12.97) | 163.0 | 173.8 | 13942 | 448.4 | 52.0 | 11080 | Cheng et al., 2019 |
| Spirogloea muscicola (CCAC 0214) | 174.0 | 171.1 | 19678 | 566 | 56.5 | 27137 | Cheng et al., 2019 |
| Closterium peracerosum-strigosum-littorale complex (NIES-67) | 365.0 | 360.0 | NA | 351* | 56.1 | 29752 | Sekimoto et al., 2023 |
| C. peracerosum-strigosum-littorale complex (NIES-68) | 361.0 | 337.0 | NA | 275* | 55.8 | 28427 | Sekimoto et al., 2023 |
| Penium margaritaceum (SAG 2640) | 4700.0 | 3661.0 | 332786 | 116.1 | 51.0 | 52333 | Jiao et al., 2020 |
NA: 数据缺失; GC含量从NCBI数据库中查得; *表示contig N50长度。
NA: Data missing; GC content retrieved from NCBI database; * indicate N50 length of the contigs.
1.1.1 Zygnematales目物种
双星藻属(Zygnema)的物种以多细胞丝状结构排列, 每个细胞包含2个叶绿体, 细胞之间的细胞壁(厚约400 nm)比外壁(约1 µm)薄得多(Feng et al., 2024)。在已测序轮藻门植物中, Z. circumcarinatum的基因组最小, 仅70 Mb左右。3个不同株系的Z. circumcarinatum基因组大小和基因数目基本相同。Z. cf. cylindricum组装的基因组为359.8 Mb, 是Z. circumcarinatum的5倍, 但其重复序列占比高达73.3% (Feng et al., 2024)。基于以上2个双星藻纲物种的全基因组信息, 通过比较基因组和共表达网络分析, 发现早期陆生植物在遗传上的新特点。例如, 植物激素几乎调节植物生长发育的所有过程, 特别是在抗逆方面(Wang et al., 2015, 2023)。在4个双星藻纲物种基因组中含有脱落酸信号转导通路的关键元件。此外, 细胞壁中的纤维素微丝是植物抵御陆地环境压力的第一道防线(Fürst-Jansen et al., 2020)。这4个基因组中含有通过基因水平转移获得与细胞壁合成相关的重要基因家族O-FucT (Feng et al., 2024)。
Mesotaenium endlicherianum为中带藻科的单细胞物种, 组装的基因组大小约173.8 Mb, 注释到11 080个基因(Cheng et al., 2019)。这些基因平均长度较长, 内含子平均长度达714.77 bp。比较演化基因组学研究发现, M. endlicherianum通过基因水平转移或其它机制, 新获得了15个直系同源群(orthogroups, OGs), 包括28个基因。扩增的直系同源群有10个, 包括67个基因。这些新获得和扩增的基因主要与植物激素信号转导及抗逆有关。值得一提的是, 通过基因水平转移, 从土壤细菌中获得了3个编码脱落酸受体基因PYR/PYL/RCAR的3个同源基因(ME000115S00012、ME000128S00179和ME000324S05315)。通过蛋白质三维结构模拟与比较, 发现水平转移获得的同源基因编码的蛋白质与拟南芥(Arabidopsis thaliana)脱落酸受体的三维结构(Nishimura et al., 2009)基本相同。由于脱落酸在植物响应环境胁迫中发挥重要作用, 特别是对干旱的响应(Fujii et al., 2009), 推测脱落酸受体基因的获得可能在植物陆地化过程中起重要作用。
1.1.2 Spirogloeales目物种
Spirogloeales目已测序物种为Spirogloea muscicola, 其基因组长度为171.1 Mb, 注释到27 137个基因(Cheng et al., 2019)。通过系统分类与比较演化基因组学分析, 证实S. muscicola是来源于一个该目新鉴定的Spirogloeophycidae科的新物种, 为生活在淡水中的单细胞淡水藻类。S. muscicola的基因数量是与其基因组大小相近的M. endlicherianum的3倍。通过全基因组共线性分析, 发现S. muscicola在近期经历了全基因组三倍化事件(whole genome triplication, WGT), 因此含有更多的基因, 尤其含有更多与抗逆(干旱和强紫外线)相关的转录因子。S. muscicola新获得和扩增的直系同源群比M. endlicherianum分别多1个, 但由于经历了全基因组三倍化事件, 新获得的直系同源群含有的基因达74个, 扩增的直系同源群含有的基因达162个。GRAS蛋白属于一类植物特有的转录因子家族, 其广泛存在于高等植物中, 参与调节多种重要的生理过程(Sun et al., 2012; Fan et al., 2021)。通过系统发生分子演化研究, 发现在S. muscicola和M. endlicherianum的基因组中都含有GRAS的同源序列, 分别为23条和8条。与PYR/PYL/RCAR相同, GRAS基因也可能是通过基因水平转移从土壤细菌中获得。全基因组重复(whole genome duplication, WGD)是生命体演化的重要驱动力, 尤其对植物在演化过程中适应剧烈的环境变化至关重要(Sémon and Wolfe, 2007)。基因水平转移能够将外源遗传材料分散到不同的谱系中, 可能为受体生物体提供新的功能或表型, 使其更好地适应环境, 因此基因水平转移也被认为是原核和真核生物演化的驱动力(Ma et al., 2022)。全基因组重复和基因水平转移赋予了S. muscicola重要的遗传创新, 为陆生植物的起源奠定了重要的遗传基础。
1.1.3 鼓藻目物种
Penium margaritaceum为单细胞藻类, 能够短暂适应长期干燥的湿地, 具有与陆地植物类似的复杂细胞壁, 可以分泌多糖黏液(Jiao et al., 2020)。P. mar- garitaceum的基因组长度为4.7 Gb (Jiao et al., 2020), 是目前已完成全基因组测序的轮藻物种中基因组最大的。与其它双星藻纲物种相比, P. margaritaceum的基因组含有更多的逆转录转座子, 这些序列与大量重复片段相关, 可能促进基因的新功能化(Jiao et al., 2020)。虽然P. margaritaceum近期未经历全基因组重复事件, 但是含有大量的重复片段。P. margaritaceum基因组新获得或扩增了与陆地化相关的基因家族和调控系统, 如调节性转录因子家族以及植物激素合成和信号转导相关的基因家族。植物细胞壁的主要成分包括嵌入到果胶和半纤维素多糖基质中的纤维素、微纤维和糖蛋白(Dehors et al., 2019)。P. margaritaceum基因组中显著扩增了与细胞壁合成相关的基因家族, 如编码糖苷水解酶(GH)、糖苷转移酶(GT)、碳水化合物酯酶(CE)和多糖裂解酶(PL)的基因家族。在P. margaritaceum中鉴定出为植物提供紫外线辐射保护的黄酮类化合物, 其基因组中含有参与黄酮类化合物合成的4-香豆酸酯: CoA连接酶(4-coumarate: coenzyme A ligase)和查尔酮合酶(chalcone synthase), 但是缺少一些关键步骤的候选基因, 表明在P. margaritaceum中可能存在与陆生植物不同的合成途径或催化酶类。因此, 结合P. margaritaceum全基因组信息和分子生物学等手段, 挖掘不同于陆生模式植物的关键代谢物质的合成途径是一个重要研究方向。
C. peracerosum-strigosum-littorale复合体由3个无法确定种间界限的Closterium属物种组成, 即C. peracerosum、C. strigosum和C. littorale。该物种复合体为纺锤形单细胞(Akatsuka et al., 2003), 拥有同配生殖系统, 即不同生殖类型的细胞从形态上无法区分(Sekimoto et al., 2023)。Sekimoto等(2023)测序并组装了该复合体的2个不同株系, 它们具有不同的交配类型(mt+和mt-)。2个株系基因组以及注释的基因数目基本一致。通过比较注释的全基因组基因, 分别鉴定到NIES-67株系和NIES-68株系的特异基因6 061个和5 610个。进而通过遗传连锁分析和生物功能验证, 确定CpMinus1基因是决定Closterium物种复合体交配型的主要因素。RWP-RK转录因子是陆生植物中氮响应和配子体发育的关键调控因子(Chardin et al., 2014)。CpMinus1基因编码蛋白的序列与陆生植物的RWP-RK转录因子聚成单系分支。与P. margaritaceum基因组中的3个同源序列相比, CpMinus1具有较长的演化支, 预示CpMinus1作为决定性别的基因似乎经历了加速演化, 这可能通过与目标DNA或蛋白互作实现(Sekimoto et al., 2023)。对物种复合体C. peracerosum-strigosum-littorale全基因测序以及其它轮藻门植物和陆生植物全基因组进行比较分析, 将为探索植物性染色体的演化提供新视角。
1.2 其它轮藻门物种全基因组
除双星藻纲外, 还有4个纲的4种轮藻已完成全基因组测序, 其中M. viride的2个不同株系分别被不同的研究组测序(表3)。相较于双星藻纲的9个物种, 轮藻门其它纲的物种在测序数量以及种类方面均较为欠缺。
表3 4个非双星藻纲的轮藻植物基因组组装统计
Table 3
| Species (strain) | Predicting genome size (Mb) | Assembly size (Mb) | Number of scaffold | Scaffold N50 (kb) | GC content (%) | Number of gene | Reference |
|---|---|---|---|---|---|---|---|
| Chara braunii (S276) | 2355.0 | 1751.5 | 11654 | 2260 | 48.3 | 23546 | Nishiyama et al., 2018 |
| Klebsormidium nitens (NIES-2285) | 117.1±21.8 | 104.0 | 1812 | 134.9 | 52.4 | 16215 | Hori et al., 2014 |
| Chlorokybus atmophyticus (CCAC 0220) | 85.0 | 74.0 | 3836 | 752.4 | 51.5 | 9066 | Wang et al., 2020 |
| Mesostigma viride (CCAC 1140) | 329.0 | 281.0 | 6924 | 113.2 | 55.0 | 9198 | Wang et al., 2020 |
| M. viride (NIES-296) | NA | 442.6 | 2363 | 2558.7 | 54.5 | 24431 | Liang et al., 2019 |
NA: 数据缺失; GC含量从NCBI数据库中查得。
NA: Data missing; GC content retrieved from NCBI database.
1.2.1 轮藻纲物种基因组
轮藻纲包含8个目, 1 223个种(Guiry, 2021; Guiry and Guiry, 2025), 但目前仅Chara braunii完成了全基因组测序(Nishiyama et al., 2018)。C. braunii为早期分化的轮藻门植物, 但在形态上比其它轮藻门植物更为复杂, 其单倍体包含1个类似茎的轴, 该轴由具有轮生的节、节间、单一的顶端分生组织和多细胞的根状体组成。C. braunii基因组大小约为2.3 Gb, 但仅有1.75 Gb被组装到scaffold水平, 注释了23 546个基因(Nishiyama et al., 2018)。内含子平均长度为 5 911.0 bp, 是M. endlicherianum的8倍, GC含量较其它轮藻低。通过比较基因组学分析, 发现C. braunii基因组具有与植物陆地化相关的遗传新特点。在其基因组中鉴定到730个转录因子或转录调控因子, 这与其复杂的形态相一致。同时鉴定到一些基因家族的特异性扩增, 如C. braunii基因组中有7个LysM-RLKs, 这可能反映出C. braunii对多种互作微生物的适应性。
1.2.2 链丝藻纲物种基因组
链丝藻纲包含3个目, 54个种, 在地球上广泛分布, 能够适应各种极端环境(Guiry, 2021; Bierenbroodspot et al., 2024; Guiry and Guiry, 2025), 目前仅Klebsormidium nitens完成了全基因组测序(Hori et al., 2014)。K. nitens为多细胞轮藻, 形态呈未分支的丝状, 除能在极端环境中生存外, 大多数适应陆地的物种也能在淡水中生存。组装到的K. nitens基因组为104 Mb, 共有16 215个基因被注释, 其基因密度较大且重复序列占比最小。通过比较基因组学分析, 发现K. nitens有1 238个蛋白与陆生植物共享, 数量多于其它绿藻植物。全基因组基因家族比较分析表明, 陆生植物基因组成员数目比较多的基因家族在K. nitens基因组中的同源基因多为单拷贝, 且这些基因主要与细胞壁的生物合成和植物激素信号转导相关。蛋白质结构域种类和组合比较分析表明, K. nitens基因组含有陆生植物中常见的90.7%蛋白结构域和84.3%结构域组合。由于基因重复和重组是植物基因组演化的动力(Kersting et al., 2012), 因此, 陆生植物中许多典型的基因或结构域可能在K. nitens的祖先中就已存在, 后来在适应与陆地生活相关的各种挑战中, 基因重复和重组最终为陆生植物提供了新的蛋白质结构域组合, 进而行使新的生物学功能。色氨酸氨基转移酶(TAA)和黄素单加氧酶(YUC)参与植物中主要的生长素生物合成途径(Mashiguchi et al., 2011)。通过同源比对和系统发生分析, 发现K. nitens基因组中编码TAA和YUC的基因通过基因水平转移获得(Wang et al., 2014)。
1.2.3 绿方藻纲物种基因组
绿方藻纲是早期分化出来的轮藻类群, 仅含1个目, 5个种(Guiry, 2021; Guiry and Guiry, 2025), 目前仅Chlorokybus atmophyticus完成了全基因组测序(Wang et al., 2020)。C. atmophyticus是亚气生或陆生多细胞藻类, 生长在土壤和石头上, 常出现在苔藓植物群落中, 具有细胞壁。其组装到的基因组大小约74.0 Mb, 注释到9 066个基因, 内含子平均长度为989 bp。在114个不同类型的转录因子和转录调控因子中, C. atmophyticus基因组中含有80个, 主要与非生物胁迫响应、发育和植物-病原体互作有关。同源异形域-亮氨酸拉链基因家族(homeodomain-leucine zipper, HD-ZIP)为植物所特有, 在生长发育中有多种功能, 主要与逆境响应相关(Ariel et al., 2007)。C. atmophyticus基因组中含4类HD-ZIP中的3类。早期具有不断进行细胞壁组分的重组和降解特性的C. atmophyticus, 细胞壁得以增强, 能灵活响应渗透压力的改变, 因更能适应快速变化的外部环境而被保留。在C. atmophyticus基因组中鉴定到的Chrsp103S00964、Chrsp134S08684和Chrsp94S08346为编码纤维素合酶(cellulose synthase, CESA/CSLD- like)基因, 但缺少编码降解木葡聚糖和木聚糖的酶和大多数果胶裂解酶基因。
1.2.4 中斑藻纲物种基因组
中斑藻纲与绿方藻纲相似, 也是早期分化的轮藻类群, 且两者互为姐妹类群。中斑藻纲仅有1个目, 2个种。目前仅Mesostigma viride的2个不同株系CCAC 1140 (Wang et al., 2020)和NIES-296 (Liang et al., 2019)完成了全基因组测序。与绿方藻纲的C. atmophyticus不同, M. viride生活在淡水中, 为单细胞藻类, 具有适合在水中运动的鞭毛, 表面覆篮子状鳞片, 但无细胞壁(Liang et al., 2019; Wang et al., 2020)。2个株系的GC含量均为55%左右, 但是基因组大小和基因数目差异较大。株系CCAC 1140组装到的基因组约281.0 Mb, 基因数为9 198。株系NIES-296组装到的基因组约442.6 Mb, 基因数为 24 431。后续将对2个株系的全基因组进行比较, 以阐明两者巨大差异的原因。在株系CCAC 1140基因组中含有80个转录因子和转录调控因子, 它们主要与非生物胁迫响应、发育和植物-病原体互作有关。在株系CCAC 1140基因组中鉴定到3个(Mesvi205S04101、Mesvi377S05585和Mesvi1458S02898)编码类纤维素合酶(cellulose synthase-like enzymes, CSLA/CSLC-like)基因, 但无编码纤维素合酶的基因(Wang et al., 2020)。不同的是, 在株系NIES-296基因组中, 鉴定到1个纤维素合酶基因和1个类纤维素合酶基因。由于2个株系的基因组均未组装到染色体水平, 都是通过同源序列分析来预测基因的生物功能, 因此后续还需在更完整的全基因组信息基础上, 结合生物化学和分子生物学实验, 验证基因功能, 最终确定M. viride基因组是否含有编码纤维素合酶的基因, 进而揭示细胞壁在绿色植物陆地化过程中的起源与演化机制。
2 轮藻门植物全基因组数据的应用
基于轮藻门植物的全基因组数据, 研究人员系统鉴定和分析了绿色植物中一些重要的基因家族和信号通路, 为绿色植物的演化以及调控重要性状的基因挖掘和功能研究提供了新思路。
2.1 转录组分析与基因挖掘
C. braunii是早期陆生植物演化和陆地化的模型。盐胁迫对水和离子运输产生重要影响, 因此与许多生物学过程相互作用, 如光合作用中的无机碳获取。基于C. braunii参考基因组(Nishiyama et al., 2018), Heß等(2023)探讨了盐胁迫对C. braunii生理和基因表达的影响, 共获得17 387个基因的表达量, 其中95个基因在盐胁迫后上调表达, 44个基因下调表达。上调表达的基因功能主要富集在离子/溶质运输和细胞壁合成, 表明这些基因在渗透适应中发挥作用; 下调表达的基因主要与光合作用和碳代谢/固定有关。C. braunii中与陆生植物早期响应脱水胁迫蛋白(early responsive to dehydration stress 4, ERD4)的编码基因在盐胁迫6小时后表达量短暂升高, 系统发生分析表明该基因起源于轮藻纲。C. braunii可在一定范围内耐受盐浓度增高, 通过不同的基因表达变化实现新的代谢稳态。因此推测这也是植物陆地化的先决条件。
植物在陆地化过程中, 面临剧烈的环境变化, 如温度和光照。Dadras等(2023)设置了42个不同温度和光照强度的组合, 对M. endlicherianum进行培养, 并对128个转录组进行测序。基于该研究的转录组信息, 将M. endlicherianum全基因组原有的11 080个基因(Cheng et al., 2019)增加到40 326个蛋白编码基因。通过加权基因共表达网络分析(weighted gene co- expression network analysis, WGCNA), 发现M. endlicherianum基因组中含有与陆生植物共享的遗传调控网络核心基因(hub gene), 如调控叶绿体发育和光反应相关核基因GLK1, 调控光形态建成的COL3和COP1。在陆生植物中, 脂滴(lipid droplet, LD)的形成与三酰甘油(triacylglycerol, TAG)的积累是应对压力反应的共同特征, 包括热、冷和干旱(Mueller et al., 2015; Gidda et al., 2016)。综合全面的转录组分析, 发现与压力依赖性脂滴形成相关的共表达模块可能在植物登陆之前就已存在。
抵抗强光照射能力是植物陆地化的关键因素。Serrano-Pérez等(2022)在高光照和低光照条件下培养K. nitens (株系NIES-2285), 并进行整合的转录组和代谢组分析。结果表明, 与低光照相比, 在高光照3小时后, 有677个基因被激活, 678个基因表达受抑制。这些差异表达的基因主要定位于叶绿体的类囊体膜, 表明叶绿体正在进行重大的重编程。被抑制的基因功能显著富集在光合作用、己糖生物合成、细胞周期和DNA代谢过程; 被激活的基因功能显著富集在核糖体生物合成、细胞质翻译启动、蛋白质折叠和氧化应激响应等过程。通过对具体基因进行功能分析, 发现K. nitens拥有快速的叶绿体逆行信号转导机制, 该转导过程可能由活性氧物质、肌醇多磷酸1-磷酸酶(inositol polyphosphate 1-phosphatase, SAL1)以及3′-磷酸腺苷-5′-磷酸(3′-phosphoadenosine-5′-phosphate, PAP)途径介导。在K. nitens基因组中, 编码SAL1的基因为kfl00096_0240。在高光强下, kfl00096_0240基因被显著抑制, 表明可能发生了PAP积累以及SAL1-PAP逆行信号转导途径的激活, 这可能是K. nitens对高光强的响应机制。
2.2 基因功能研究
基于轮藻门植物的全基因组信息, 众多研究对植物激素信号通路关键基因、转录因子和细胞壁合成相关基因进行预测和系统发生分析。但通过整合生物化学和分子生物学进行基因功能研究较少。
生长素梯度在很大程度上依赖其定向(极性)的细胞间运输, 这一过程由不对称分布的质膜定位的PIN跨膜生长素外排转运蛋白介导(Adamowski and Friml, 2015)。Skokan等(2019)对K. nitens基因组中唯一的PIN同源基因kfPIN进行功能研究。将kfPIN在拟南芥和小立碗藓(Physcomitrella patens)中过表达, 结果均引起生长反应, 表明生长素稳态发生变化。在拟南芥根毛中异位表达kfPIN能够抑制根毛生长, 表明kfPIN具有促进植物生长素运输的能力。亚细胞定位分析发现, kfPIN在拟南芥中主要定位于质膜。进一步通过免疫染色实验发现, 与陆生植物不同, kfPIN不能在K. nitens中极性定位。其在K. nitens中的定位模式主要位于细胞外围并在侧面富集, 而非集中在细胞与细胞间的接触界面, 这提示其可能介导生长素从细胞内外排, 因此推测这可能是kfPIN的原始功能。通过一系列生物学实验, 已确定了PIN同源基因kfPIN的生物学功能, 发现了PIN基因的原始功能, 为深入理解绿色植物适应环境的机制提供了新见解。
3 总结与展望
从2014年第1个轮藻基因组被测序, 到目前已有5个纲10个种完成了全基因组测序。覆盖物种种类多样全面的高质量参考基因组对于功能基因的挖掘、物种分类和重要生物机制的阐明具有重要意义(宫少达等, 2024)。基于轮藻门植物的全基因组信息, 已基本确定双星藻纲是与陆生植物亲缘关系最近的姐妹系, 而不是表型复杂的轮藻纲。这显示了全基因组数据对物种进化树构建的重要性。在明确轮藻门与陆生植物系统发生关系的基础上, 发现了一系列扩张的基因家族, 如转录因子和植物激素信号转导关键元件。基因水平转移在轮藻门植物预适应陆地环境中发挥重要作用。一系列新发现为深入理解绿色植物从水生环境到适应陆地环境的分子机制奠定了基础。与水生环境相比, 陆地环境更加干燥, 光强和氧气浓度更高, 温度变化更加剧烈(Horňák, 2022)。因此, 作为陆生植物的姐妹系, 轮藻门植物对陆地环境预适应机制有望应用于作物的遗传改良, 如抗旱和抗盐碱(Simmons and Herman, 2023 )。
相对于轮藻门已知的5 761个物种(Guiry, 2021; Guiry and Guiry, 2025), 目前仅10个种完成了全基因组测序, 占总数的0.17%。这10个已测序物种分布在5个纲, 鞘毛藻纲目前尚无测序物种。除测序物种分布不均衡外, 全基因组测序质量也有待提高。除Z. circumcarinatum的SAG 698-1b株系的基因组达到染色体水平, 其它轮藻基因组都仅组装到scaffold水平, 全基因组信息均有不同程度的缺失(表1, 表2), 这严重阻碍了比较基因组学和重要遗传资源的挖掘。随着三代测序技术和组装算法的不断发展, 尤其是高准确性的HiFi和高连续性的ONT三代测序技术的应用, 基因组组装进入了新阶段(宫少达等, 2024; 王英豪等, 2024)。因此, 未来应将新技术和算法应用于轮藻门植物基因组染色体水平的组装, 甚至是端粒到端粒(telomere to telomere, T2T)水平的组装。
此外, 由于单一个体的参考基因组不能代表整个物种的遗传多样性, 因此将多个个体的参考基因组整合为该物种的泛基因组(pangenome), 以此代表全部基因组信息的集合, 将成为揭示生物体丰富的遗传变异和环境适应机制的重要手段(Huang et al., 2023)。但目前尚未见从泛基因组水平对轮藻门植物进行遗传多样性研究。仅有2个研究组对M. viride的2个不同株系进行了全基因测序, 发现二者基因组大小有显著差异(Liang et al., 2019; Wang et al., 2020)。对Z. circumcarinatum的3个不同株系(SAG 698-1b、UTEX 1559和UTEX 1560)进行了全基因组测序, 显示它们的基因组大小和GC含量基本一致(Feng et al., 2024)。后续可考虑对轮藻门植物的不同亚种或株系进行测序和组装, 在泛基因组水平系统研究轮藻门植物的遗传多样性和环境适应机制, 并挖掘重要遗传资源。
通过演化比较基因组学研究, 现已在轮藻门植物全基因组中预测和分析了在绿色植物陆地化过程中的重要基因(Horňák, 2022)。但序列的同源性并不一定表明功能上的保守性(薛进庄等, 2022)。因此, 在全基因组信息基础上, 对预测具有重要生物功能的轮藻门植物基因进行系统完整的生物化学、分子生物学和遗传学的实验验证十分必要。例如, K. nitens基因组中仅有的PIN同源基因kfPIN虽具有运输植物生长素的功能, 但与陆生植物中典型的PIN基因在定位模式上不同(Skokan et al., 2019)。生物学实验不仅验证了其功能, 还发现了不同于传统模式植物中的生物学新特性。因此, 整合轮藻门植物基因组信息和开展生物学实验, 将有助于挖掘新的非模式功能基因, 同时拓宽对生物适应性和调控模式的认识。
作者贡献声明
夏琳凤: 文献搜集整理, 作图和文章初稿撰写; 李瑞: 参与文章修改和校对; 王海政: 数据整理和图片校对; 冯大领: 指导并提出文章修改建议; 王春阳: 选题构思和修改文章, 提供资金支持。
通讯作者/团队简介
王春阳, 河北农业大学生命科学学院副教授, 硕士生导师。主要从事植物演化基因组学和功能基因组学研究。以第一作者或通讯作者身份在Molecular Plant、Trends in Plant Science及Plant Physiology等期刊发表论文近10篇。目前其研究团队主要从事绿色植物演化基因组学研究; 同时, 以白菜类作物为模式植物, 利用比较基因组学和群体遗传学以及分子生物学等手段, 开发多组学数据整合分析方法流程, 挖掘白菜类作物重要农艺性状的遗传调控位点。
参考文献

When mating-type plus (mt+) and minus (mt-) cells of the Closterium peracerosum-strigosum-littorale complex were mixed in nitrogen-depleted mating medium, secretion of mucilage containing uronic acid from cells was markedly activated and the mucilage accumulated around the cells. Substances with the ability to stimulate mucilage secretion from mt+ and mt- cells were detected in media in which mt- and mt+ cells had been separately cultured, respectively. We designated the active substances secreted from mt+ and mt- cells mucilage secretion-stimulating pheromone (MS-SP)-plus and MS-SP-minus, respectively. Activity of MS-SP-plus and MS-SP-minus decreased to 20% level by incubation at 80 degrees C for 10 min. Light was indispensable for the secretion of mucilage. The secretion of MS-SP-plus and MS-SP-minus decreased with dark treatment. MS-SP-plus eluted at around 95 k from a gel filtration column, and reacted with antibodies against two subunits of protoplast-release-inducing protein (PR-IP), which induces protoplast release from mt- cells. MS-SP-minus eluted at around 20 k from a gel filtration column, and reacted with an antibody against the PR-IP inducer, which induces the secretion of PR-IP from mt+ cells. In addition, purified PR-IP and PR-IP inducer stimulated mucilage secretion from mt- and mt+ cells, respectively. These results strongly suggested that MS-SP-plus and MS-SP-minus were the same molecules as the PR-IP and the PR-IP inducer, respectively.
The HD-Zip family of transcription factors is unique to the plant kingdom. These proteins exhibit the singular combination of a homeodomain with a leucine zipper acting as a dimerization motif. They can be classified into four subfamilies, according to a set of distinctive features that include DNA-binding specificities, gene structures, additional common motifs and physiological functions. Some HD-Zip proteins participate in organ and vascular development or meristem maintenance. Others mediate the action of hormones or are involved in responses to environmental conditions. Here, we review recent data for this family of transcription factors from a wide variety of plant species to unravel their crucial role in plant development.
The plant specific RWP-RK family of transcription factors, initially identified in legumes and Chlamydomonas, are found in all vascular plants, green algae, and slime molds. These proteins possess a characteristic RWP-RK motif, which mediates DNA binding. Based on phylogenetic and domain analyses, we classified the RWP-RK proteins of six different species in two subfamilies: the NIN-like proteins (NLPs), which carry an additional PB1 domain at their C-terminus, and the RWP-RK domain proteins (RKDs), which are divided into three subgroups. Although, the functional analysis of this family is still in its infancy, several RWP-RK proteins have a key role in regulating responses to nitrogen availability. The nodulation-specific NIN proteins are involved in nodule organogenesis and rhizobial infection under nitrogen starvation conditions. Arabidopsis NLP7 in particular is a major player in the primary nitrate response. Several RKDs act as transcription factors involved in egg cell specification and differentiation or gametogenesis in algae, the latter modulated by nitrogen availability. Further studies are required to extend the general picture of the functional role of these exciting transcription factors. © The Author 2014. Published by Oxford University Press on behalf of the Society for Experimental Biology. All rights reserved. For permissions, please email: journals.permissions@oup.com.
The transition to a terrestrial environment, termed terrestrialization, is generally regarded as a pivotal event in the evolution and diversification of the land plant flora that changed the surface of our planet. Through phylogenomic studies, a group of streptophyte algae, the Zygnematophyceae, have recently been recognized as the likely sister group to land plants (embryophytes). Here, we report genome sequences and analyses of two early diverging Zygnematophyceae (Spirogloea muscicola gen. nov. and Mesotaenium endlicherianum) that share the same subaerial/terrestrial habitat with the earliest-diverging embryophytes, the bryophytes. We provide evidence that genes (i.e., GRAS and PYR/PYL/RCAR) that increase resistance to biotic and abiotic stresses in land plants, in particular desiccation, originated or expanded in the common ancestor of Zygnematophyceae and embryophytes, and were gained by horizontal gene transfer (HGT) from soil bacteria. These two Zygnematophyceae genomes represent a cornerstone for future studies to understand the underlying molecular mechanism and process of plant terrestrialization.Crown Copyright © 2019. Published by Elsevier Inc. All rights reserved.
Plant terrestrialization brought forth the land plants (embryophytes). Embryophytes account for most of the biomass on land and evolved from streptophyte algae in a singular event. Recent advances have unravelled the first full genomes of the closest algal relatives of land plants; among the first such species was Mesotaenium endlicherianum. Here we used fine-combed RNA sequencing in tandem with a photophysiological assessment on Mesotaenium exposed to a continuous range of temperature and light cues. Our data establish a grid of 42 different conditions, resulting in 128 transcriptomes and ~1.5 Tbp (~9.9 billion reads) of data to study the combinatory effects of stress response using clustering along gradients. Mesotaenium shares with land plants major hubs in genetic networks underpinning stress response and acclimation. Our data suggest that lipid droplet formation and plastid and cell wall-derived signals have denominated molecular programmes since more than 600 million years of streptophyte evolution-before plants made their first steps on land.© 2023. The Author(s).
Contents Summary 1428 I. The singularity of plant terrestrialization 1428 II. Adaptation vs exaptation - what shaped the land plant toolkit? 1430 III. Trait mosaicism in (higher-branching) streptophyte algae 1431 IV.a streptophyte algal perspective on land plant trait evolution 1432 Acknowledgements 1432 ORCID 1433 References 1433 SUMMARY: Photosynthetic eukaryotes thrive anywhere there is sunlight and water. But while such organisms are exceptionally diverse in form and function, only one phototrophic lineage succeeded in rising above its substrate: the land plants (embryophytes). Molecular phylogenetic data show that land plants evolved from streptophyte algae most closely related to extant Zygnematophyceae, and one of the principal aims of plant evolutionary biology is to uncover the key features of such algae that enabled this important transition. At the present time, however, mosaic and reductive evolution blur our picture of the closest algal ancestors of plants. Here we discuss recent progress and problems in inferring the biology of the algal progenitor of the terrestrial photosynthetic macrobiome.© 2018 The Authors. New Phytologist © 2018 New Phytologist Trust.
During evolution of land plants, the first colonizing species presented leafy-dominant gametophytes, found in non-vascular plants (bryophytes). Today, bryophytes include liverworts, mosses, and hornworts. In the first seedless vascular plants (lycophytes), the sporophytic stage of life started to be predominant. In the seed producing plants, gymnosperms and angiosperms, the gametophytic stage is restricted to reproduction. In mosses and ferns, the haploid spores germinate and form a protonema, which develops into a leafy gametophyte producing rhizoids for anchorage, water and nutrient uptakes. The basal gymnosperms (cycads and ) reproduce by zooidogamy. Their pollen grains develop a multi-branched pollen tube that penetrates the nucellus and releases flagellated sperm cells that swim to the egg cell. The pollen grain of other gymnosperms (conifers and gnetophytes) as well as angiosperms germinates and produces a pollen tube that directly delivers the sperm cells to the ovule (siphonogamy). These different gametophytes, which are short or long-lived structures, share a common tip-growing mode of cell expansion. Tip-growth requires a massive cell wall deposition to promote cell elongation, but also a tight spatial and temporal control of the cell wall remodeling in order to modulate the mechanical properties of the cell wall. The growth rate of these cells is very variable depending on the structure and the species, ranging from very slow (protonemata, rhizoids, and some gymnosperm pollen tubes), to a slow to fast-growth in other gymnosperms and angiosperms. In addition, the structural diversity of the female counterparts in angiosperms (dry, semi-dry wet stigmas, short long, solid hollow styles) will impact the speed and efficiency of sperm delivery. As the evolution and diversity of the cell wall polysaccharides accompanied the diversification of cell wall structural proteins and remodeling enzymes, this review focuses on our current knowledge on the biochemistry, the distribution and remodeling of the main cell wall polymers (including cellulose, hemicelluloses, pectins, callose, arabinogalactan-proteins and extensins), during the tip-expansion of gametophytes from bryophytes, pteridophytes (lycophytes and monilophytes), gymnosperms and the monocot and eudicot angiosperms.
Charophytes represent a diverse assemblage of extant green algae that are the sister lineage to land plants. 500-600+ million years ago, a charophyte progenitor successfully colonized land and subsequently gave rise to land plants. Charophytes have diverse but relatively simple body plans that make them highly attractive organisms for many areas of biological research. At the cellular level, many charophytes have been used for deciphering cytoskeletal networks and their dynamics, membrane trafficking, extracellular matrix secretion and cell division mechanisms. Some charophytes live in challenging habitats and have become excellent models for elucidating the cellular and molecular effects of various abiotic stressors on plant cells. Recent sequencing of several charophyte genomes has also opened doors for the dissection of biosynthetic and signaling pathways. While we are only in an infancy stage in elucidating the cell biology of charophytes, the future application of novel analytical methodologies in charophyte studies that include a broader survey of inclusive taxa will enhance our understanding of plant evolution and cell dynamics.© The Author(s) 2022. Published by Oxford University Press on behalf of American Society of Plant Biologists.
Embryophytes (land plants) can be found in almost any habitat on the Earth's surface. All of this ecologically diverse embryophytic flora arose from algae through a singular evolutionary event. Traits that were, by their nature, indispensable for the singular conquest of land by plants were those that are key for overcoming terrestrial stressors. Not surprisingly, the biology of land plant cells is shaped by a core signaling network that connects environmental cues, such as stressors, to the appropriate responses-which, thus, modulate growth and physiology. When did this network emerge? Was it already present when plant terrestrialization was in its infancy? A comparative approach between land plants and their algal relatives, the streptophyte algae, allows us to tackle such questions and resolve parts of the biology of the earliest land plants. Exploring the biology of the earliest land plants might shed light on exactly how they overcame the challenges of terrestrialization. Here, we outline the approaches and rationale underlying comparative analyses towards inferring the genetic toolkit for the stress response that aided the earliest land plants in their conquest of land.© The Author(s) 2020. Published by Oxford University Press on behalf of the Society for Experimental Biology.
Eukaryotic cells compartmentalize neutral lipids into organelles called lipid droplets (LDs), and while much is known about the role of LDs in storing triacylglycerols in seeds, their biogenesis and function in nonseed tissues are poorly understood. Recently, we identified a class of plant-specific, lipid droplet-associated proteins (LDAPs) that are abundant components of LDs in nonseed cell types. Here, we characterized the three LDAPs in Arabidopsis (Arabidopsis thaliana) to gain insight to their targeting, assembly, and influence on LD function and dynamics. While all three LDAPs targeted specifically to the LD surface, truncation analysis of LDAP3 revealed that essentially the entire protein was required for LD localization. The association of LDAP3 with LDs was detergent sensitive, but the protein bound with similar affinity to synthetic liposomes of various phospholipid compositions, suggesting that other factors contributed to targeting specificity. Investigation of LD dynamics in leaves revealed that LD abundance was modulated during the diurnal cycle, and characterization of LDAP misexpression mutants indicated that all three LDAPs were important for this process. LD abundance was increased significantly during abiotic stress, and characterization of mutant lines revealed that LDAP1 and LDAP3 were required for the proper induction of LDs during heat and cold temperature stress, respectively. Furthermore, LDAP1 was required for proper neutral lipid compartmentalization and triacylglycerol degradation during postgerminative growth. Taken together, these studies reveal that LDAPs are required for the maintenance and regulation of LDs in plant cells and perform nonredundant functions in various physiological contexts, including stress response and postgerminative growth.© 2016 American Society of Plant Biologists. All Rights Reserved.
The orange subfamily (Aurantioideae) contains several Citrus species cultivated worldwide, such as sweet orange and lemon. The origin of Citrus species has long been debated and less is known about the Aurantioideae. Here, we compiled the genome sequences of 314 accessions, de novo assembled the genomes of 12 species and constructed a graph-based pangenome for Aurantioideae. Our analysis indicates that the ancient Indian Plate is the ancestral area for Citrus-related genera and that South Central China is the primary center of origin of the Citrus genus. We found substantial variations in the sequence and expression of the PH4 gene in Citrus relative to Citrus-related genera. Gene editing and biochemical experiments demonstrate a central role for PH4 in the accumulation of citric acid in citrus fruits. This study provides insights into the origin and evolution of the orange subfamily and a regulatory mechanism underpinning the evolution of fruit taste.© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
The embryophytes (land plants) have long been thought to be related to the green algal group Charophyta, though the nature of this relationship and the origin of the land plants have remained unresolved. A four-gene phylogenetic analysis was conducted to investigate these relationships. This analysis supports the hypothesis that the land plants are placed phylogenetically within the Charophyta, identifies the Charales (stoneworts) as the closest living relatives of plants, and shows the Coleochaetales as sister to this Charales/land plant assemblage. The results also support the unicellular flagellate Mesostigma as the earliest branch of the charophyte lineage. These findings provide insight into the nature of the ancestor of plants, and have broad implications for understanding the transition from aquatic green algae to terrestrial plants.
Plant genomes are generally very large, mostly paleopolyploid, and have numerous gene duplicates and complex genomic features such as repeats and transposable elements. Many of these features have been hypothesized to enable plants, which cannot easily escape environmental challenges, to rapidly adapt. Another mechanism, which has recently been well described as a major facilitator of rapid adaptation in bacteria, animals, and fungi but not yet for plants, is modular rearrangement of protein-coding genes. Due to the high precision of profile-based methods, rearrangements can be well captured at the protein level by characterizing the emergence, loss, and rearrangements of protein domains, their structural, functional, and evolutionary building blocks. Here, we study the dynamics of domain rearrangements and explore their adaptive benefit in 27 plant and 3 algal genomes. We use a phylogenomic approach by which we can explain the formation of 88% of all arrangements by single-step events, such as fusion, fission, and terminal loss of domains. We find many domains are lost along every lineage, but at least 500 domains are novel, that is, they are unique to green plants and emerged more or less recently. These novel domains duplicate and rearrange more readily within their genomes than ancient domains and are overproportionally involved in stress response and developmental innovations. Novel domains more often affect regulatory proteins and show a higher degree of structural disorder than ancient domains. Whereas a relatively large and well-conserved core set of single-domain proteins exists, long multi-domain arrangements tend to be species-specific. We find that duplicated genes are more often involved in rearrangements. Although fission events typically impact metabolic proteins, fusion events often create new signaling proteins essential for environmental sensing. Taken together, the high volatility of single domains and complex arrangements in plant genomes demonstrate the importance of modularity for environmental adaptability of plants.
How horizontal gene transfer (HGT) has contributed to the evolution of animals and plants remains a major puzzle. Despite recent progress, defining the overall scale and pattern of HGT events in land plants has been largely elusive. In this study, we performed systematic analyses for acquired genes in different plant groups and throughout land plant evolution. We found that relatively recent HGT events occurred in charophytes and all major land plant groups, but their frequency declined rapidly in seed plants. Two major episodes of HGT events occurred in land plant evolution, corresponding to the early evolution of streptophytes and the origin of land plants, respectively. Importantly, a vast majority of the genes acquired in the two episodes have been retained in descendant groups, affecting numerous activities and processes of land plants. We analyzed some of the acquired genes involved in stress responses, ion and metabolite transport, growth and development, and specialized metabolism, and further assessed the cumulative effects of HGT in land plants.Copyright © 2022 The Author. Published by Elsevier Inc. All rights reserved.
The phytohormone auxin plays critical roles in the regulation of plant growth and development. Indole-3-acetic acid (IAA) has been recognized as the major auxin for more than 70 y. Although several pathways have been proposed, how auxin is synthesized in plants is still unclear. Previous genetic and enzymatic studies demonstrated that both TRYPTOPHAN AMINOTRANSFERASE OF ARABIDOPSIS (TAA) and YUCCA (YUC) flavin monooxygenase-like proteins are required for biosynthesis of IAA during plant development, but these enzymes were placed in two independent pathways. In this article, we demonstrate that the TAA family produces indole-3-pyruvic acid (IPA) and the YUC family functions in the conversion of IPA to IAA in Arabidopsis (Arabidopsis thaliana) by a quantification method of IPA using liquid chromatography-electrospray ionization-tandem MS. We further show that YUC protein expressed in Escherichia coli directly converts IPA to IAA. Indole-3-acetaldehyde is probably not a precursor of IAA in the IPA pathway. Our results indicate that YUC proteins catalyze a rate-limiting step of the IPA pathway, which is the main IAA biosynthesis pathway in Arabidopsis.
The phytohormone abscisic acid (ABA) acts in seed dormancy, plant development, drought tolerance, and adaptive responses to environmental stresses. Structural mechanisms mediating ABA receptor recognition and signaling remain unknown but are essential for understanding and manipulating abiotic stress resistance. Here, we report structures of pyrabactin resistance 1 (PYR1), a prototypical PYR/PYR1-like (PYL)/regulatory component of ABA receptor (RCAR) protein that functions in early ABA signaling. The crystallographic structure reveals an alpha/beta helix-grip fold and homodimeric assembly, verified in vivo by coimmunoprecipitation. ABA binding within a large internal cavity switches structural motifs distinguishing ABA-free "open-lid" from ABA-bound "closed-lid" conformations. Small-angle x-ray scattering suggests that ABA signals by converting PYR1 to a more compact, symmetric closed-lid dimer. Site-directed PYR1 mutants designed to disrupt hormone binding lose ABA-triggered interactions with type 2C protein phosphatase partners in planta.
Land plants evolved from charophytic algae, among which Charophyceae possess the most complex body plans. We present the genome of Chara braunii; comparison of the genome to those of land plants identified evolutionary novelties for plant terrestrialization and land plant heritage genes. C. braunii employs unique xylan synthases for cell wall biosynthesis, a phragmoplast (cell separation) mechanism similar to that of land plants, and many phytohormones. C. braunii plastids are controlled via land-plant-like retrograde signaling, and transcriptional regulation is more elaborate than in other algae. The morphological complexity of this organism may result from expanded gene families, with three cases of particular note: genes effecting tolerance to reactive oxygen species (ROS), LysM receptor-like kinases, and transcription factors (TFs). Transcriptomic analysis of sexual reproductive structures reveals intricate control by TFs, activity of the ROS gene network, and the ancestral use of plant-like storage and stress protection proteins in the zygote.Copyright © 2018 Elsevier Inc. All rights reserved.
The evolutionary emergence of land plant body plans transformed the planet. However, our understanding of this formative episode is mired in the uncertainty associated with the phylogenetic relationships among bryophytes (hornworts, liverworts, and mosses) and tracheophytes (vascular plants). Here we attempt to clarify this problem by analyzing a large transcriptomic dataset with models that allow for compositional heterogeneity between sites. Zygnematophyceae is resolved as sister to land plants, but we obtain several distinct relationships between bryophytes and tracheophytes. Concatenated sequence analyses that can explicitly accommodate site-specific compositional heterogeneity give more support for a mosses-liverworts clade, "Setaphyta," as the sister to all other land plants, and weak support for hornworts as the sister to all other land plants. Bryophyte monophyly is supported by gene concatenation analyses using models explicitly accommodating lineage-specific compositional heterogeneity and analyses of gene trees. Both maximum-likelihood analyses that compare the fit of each gene tree to proposed species trees and Bayesian supertree estimation based on gene trees support bryophyte monophyly. Of the 15 distinct rooted relationships for embryophytes, we reject all but three hypotheses, which differ only in the position of hornworts. Our results imply that the ancestral embryophyte was more complex than has been envisaged based on topologies recognizing liverworts as the sister lineage to all other embryophytes. This requires many phenotypic character losses and transformations in the liverwort lineage, diminishes inconsistency between phylogeny and the fossil record, and prompts re-evaluation of the phylogenetic affinity of early land plant fossils, the majority of which are considered stem tracheophytes.Copyright © 2018 The Author(s). Published by Elsevier Ltd.. All rights reserved.
Background: Next-generation sequencing has provided a wealth of plastid genome sequence data from an increasingly diverse set of green plants (Viridiplantae). Although these data have helped resolve the phylogeny of numerous clades (e. g., green algae, angiosperms, and gymnosperms), their utility for inferring relationships across all green plants is uncertain. Viridiplantae originated 700-1500 million years ago and may comprise as many as 500,000 species. This clade represents a major source of photosynthetic carbon and contains an immense diversity of life forms, including some of the smallest and largest eukaryotes. Here we explore the limits and challenges of inferring a comprehensive green plant phylogeny from available complete or nearly complete plastid genome sequence data. Results: We assembled protein-coding sequence data for 78 genes from 360 diverse green plant taxa with complete or nearly complete plastid genome sequences available from GenBank. Phylogenetic analyses of the plastid data recovered well-supported backbone relationships and strong support for relationships that were not observed in previous analyses of major subclades within Viridiplantae. However, there also is evidence of systematic error in some analyses. In several instances we obtained strongly supported but conflicting topologies from analyses of nucleotides versus amino acid characters, and the considerable variation in GC content among lineages and within single genomes affected the phylogenetic placement of several taxa. Conclusions: Analyses of the plastid sequence data recovered a strongly supported framework of relationships for green plants. This framework includes: i) the placement of Zygnematophyceace as sister to land plants (Embryophyta), ii) a clade of extant gymnosperms (Acrogymnospermae) with cycads + Ginkgo sister to remaining extant gymnosperms and with gnetophytes (Gnetophyta) sister to non-Pinaceae conifers (Gnecup trees), and iii) within the monilophyte clade (Monilophyta), Equisetales + Psilotales are sister to Marattiales + leptosporangiate ferns. Our analyses also highlight the challenges of using plastid genome sequences in deep-level phylogenomic analyses, and we provide suggestions for future analyses that will likely incorporate plastid genome sequence data for thousands of species. We particularly emphasize the importance of exploring the effects of different partitioning and character coding strategies.
Polyploidy has been widely appreciated as an important force in the evolution of plant genomes, but now it is recognized as a common phenomenon throughout eukaryotic evolution. Insight into this process has been gained by analyzing the plant, animal, fungal, and recently protozoan genomes that show evidence of whole genome duplication (a transient doubling of the entire gene repertoire of an organism). Moreover, comparative analyses are revealing the evolutionary processes that occur as multiple related genomes diverge from a shared polyploid ancestor, and in individual genomes that underwent several successive rounds of duplication. Recent research including laboratory studies on synthetic polyploids indicates that genome content and gene expression can change quickly after whole genome duplication and that cross-genome regulatory interactions are important. We have a growing understanding of the relationship between whole genome duplication and speciation. Further, recent studies are providing insights into why some gene pairs survive in duplicate, whereas others do not.
PIN-FORMED (PIN) transporters mediate directional, intercellular movement of the phytohormone auxin in land plants. To elucidate the evolutionary origins of this developmentally crucial mechanism, we analysed the single PIN homologue of a simple green alga Klebsormidium flaccidum. KfPIN functions as a plasma membrane-localized auxin exporter in land plants and heterologous models. While its role in algae remains unclear, PIN-driven auxin export is probably an ancient and conserved trait within streptophytes.
Plant hormones modulate plant growth, development, and defense. However, many aspects of the origin and evolution of plant hormone signaling pathways remain obscure. Here, we use a comparative genomic and phylogenetic approach to investigate the origin and evolution of nine major plant hormone (abscisic acid, auxin, brassinosteroid, cytokinin, ethylene, gibberellin, jasmonate, salicylic acid, and strigolactone) signaling pathways. Our multispecies genome-wide analysis reveals that: (1) auxin, cytokinin, and strigolactone signaling pathways originated in charophyte lineages; (2) abscisic acid, jasmonate, and salicylic acid signaling pathways arose in the last common ancestor of land plants; (3) gibberellin signaling evolved after the divergence of bryophytes from land plants; (4) the canonical brassinosteroid signaling originated before the emergence of angiosperms but likely after the split of gymnosperms and angiosperms; and (5) the origin of the canonical ethylene signaling pathway postdates shortly the emergence of angiosperms. Our findings might have important implications in understanding the molecular mechanisms underlying the emergence of land plants. © 2015 American Society of Plant Biologists. All Rights Reserved.
Mounting evidence suggests that terrestrialization of plants started in streptophyte green algae, favoured by their dual existence in freshwater and subaerial/terrestrial environments. Here, we present the genomes of Mesostigma viride and Chlorokybus atmophyticus, two sister taxa in the earliest-diverging clade of streptophyte algae dwelling in freshwater and subaerial/terrestrial environments, respectively. We provide evidence that the common ancestor of M. viride and C. atmophyticus (and thus of streptophytes) had already developed traits associated with a subaerial/terrestrial environment, such as embryophyte-type photorespiration, canonical plant phytochrome, several phytohormones and transcription factors involved in responses to environmental stresses, and evolution of cellulose synthase and cellulose synthase-like genes characteristic of embryophytes. Both genomes differed markedly in genome size and structure, and in gene family composition, revealing their dynamic nature, presumably in response to adaptations to their contrasting environments. The ancestor of M. viride possibly lost several genomic traits associated with a subaerial/terrestrial environment following transition to a freshwater habitat.
The transition of the algal ancestor of land plants from fresh water to land is important in the study of plant evolutionarybiology, which has also changed the terrestrial ecosystem of the earth but the evolutionary mechanism of plant terrestrializationremains unknown. The recent report of high-quality reference genomes of several streptophyte algae and one hornwort sheds light onthis major event. Based on these studies, we can draw the following conclusions: ①Zygnematophyceae was found to be the closestsister group to the common ancestor of land plants found so far; ②The acquisition of GRAS and PYL genes from soil bacteria viahorizontal gene transfer plays an important role in driving the adaptive process of plant terrestrialization; ③The terrestrialization ofplant ancestors is an asymptotic evolutionary process, and the genetic material has gone through a series of pre-adaptation processes,which many gene families previously thought to be unique to land plants exist in more primitive green algae; ④Genomics is animportant method for studying terrestrialization and its reversibility process (such as some aquatic angiosperms). The molecularmechanism of the adaptive process of plant terrestrialization is still one of the current research hotspots, including artificial designof “experimental evolution”, horizontal gene transfer, a natural transgenic engineering event, and its key role in plant evolution andadaptation.
Plant genomes provide essential and vital basic resources for studying many aspects of plant biology and applications (for example, breeding). From 2000 to 2020, 1,144 genomes of 782 plant species were sequenced. In the past three years (2021-2023), 2,373 genomes of 1,031 plant species, including 793 newly sequenced species, have been assembled, representing a great leap. The 2,373 newly assembled genomes, of which 63 are telomere-to-telomere assemblies and 921 have been generated in pan-genome projects, cover the major phylogenetic clades. Substantial advances in read length, throughput, accuracy and cost-effectiveness have notably simplified the achievement of high-quality assemblies. Moreover, the development of multiple software tools using different algorithms offers the opportunity to generate more complete and complex assemblies. A database named N3: plants, genomes, technologies has been developed to accommodate the metadata associated with the 3,517 genomes that have been sequenced from 1,575 plant species since 2000. We also provide an outlook for emerging opportunities in plant genome sequencing.© 2024. Springer Nature Limited.
The class of conjugating green algae, Zygnematophyceae (Conjugatophyceae), is extremely rich in species and has attracted the interest of phycologists for a long time. It is now widely accepted that this class of charophyte algae holds a key position in the phylogenetic tree of streptophytes, where they represent the closest relatives to all land plants (embryophytes). It is increasingly evident that robust model plants that can be easily cultivated and genetically transformed are necessary to better understand the process of terrestrialization and the related molecular, cellular, and physiological adaptations. Living algae collections play an important role, not only for phylogenomic-based taxonomy but also for screening for suitable model organisms. For this review, we screened six major public algae collections for Zygnematophyceae strains and established a cumulative list comprising 738 different taxa (including species, subspecies, varieties, and forms). From the described biodiversity with 8883 registered taxa (AlgaeBase) the cultured Zygnematophyceae taxa worldwide cover only ~8.3%. We review the past research on this clade of algae and discuss it from the perspective of establishing a model organism. We present data on the life cycle of the genera Micrasterias and Spirogyra, representing the orders Desmidiales and Zygnematales, and outline the current status of genetic transformation of Zygnematophyceae algae and future research perspectives.© The Author(s) 2020. Published by Oxford University Press on behalf of the Society for Experimental Biology. All rights reserved. For permissions, please email: journals.permissions@oup.com.

{kind=link}
{kind=link}