
 首页
首页

近20年来, 基因组研究蓬勃发展(Sun et al., 2022b; Kress et al., 2022)。测序技术和生物信息技术的不断推新升级, 使得基因组、转录组、蛋白质组和代谢组等多组学联合分析成为可能, 极大地推进了植物生物学各领域的研究(谢玲娟等, 2021; Sun et al., 2022a; Kress et al., 2022)。不同植物基因组因其特性不同成为探索特定生物学问题的重要基础(Sun et al., 2022b)。Zhang等(2020)通过对2个榕属(Ficus)基因组和1个传粉榕小蜂(Eupristina verticillata)的基因组进行比较分析, 探讨了气生根、雌雄同株及雌雄异株的起源, 为揭示动、植物协同进化的分子机制提供了重要资料。藻苔(Takakia lepidozioides)、攀枝花苏铁(Cycas panzhihuaensis)、油松(Pinus tabuliformis)、百岁兰(Welwitschia mirabilis)和无油樟(Amborella trichopoda)等基因组的测序不仅为研究陆地植物起源和演化提供了重要依据, 还为进一步研究积累了数据(Amborella Genome Project, 2013; Wan et al., 2021; Liu et al., 2022; Niu et al., 2022; Hu et al., 2023b)。通过对水稻(Oryza sativa) (Jing et al., 2023)、葡萄(Vitis vinifera) (Dong et al., 2023)和西瓜(Citrullus lanatus) (Wu et al., 2023)等农作物的基因组研究发掘出与品质相关的重要基因, 使人们更关注作物的起源和驯化。科学家提出了全部真核生物的测序计划——地球生物基因组计划(The Earth Bio-Genome Project), 目的是提升和重塑人们对生物学、生态系统和进化的理解, 促进生物多样性的保存、保护和再生, 并最大限度地提高基因组资料对社会和人类的回报(Lewin et al., 2018; Blaxter et al., 2022)。
随着测序技术的进步、测序成本的降低以及全球经济的发展, 全基因组研究有加速的趋势。自第1个植物基因组序列被报道(The Arabidopsis Genome Initiative, 2000)以来, 截至2020年底, 已有788个植物物种的1 031个基因组被测序和报道(Sun et al., 2022b)。其中农作物的基因组测序受到更多的关注, 已测序物种最多的禾本科(Poaceae)、十字花科(Brassicaceae)和豆科(Fabaceae)均为重要的经济作物科(Sun et al., 2022b)。虽然林木物种基因组研究快速发展, 已测序乔木物种达218种, 占已测序维管植物的29% (图1A) (谢玲娟等, 2021), 而许多木本植物的大科尚未得到足够重视。在全球最大的5个木本植物科中(Beech et al., 2017), 仅有桃金娘科(Myrtaceae)已测序24个乔木物种, 为已测序乔木最多的科, 豆科、茜草科(Rubiaceae)、樟科(Lauraceae)和大戟科(Euphorbiaceae)仅有几个代表物种完成测序(图1B)。
图1
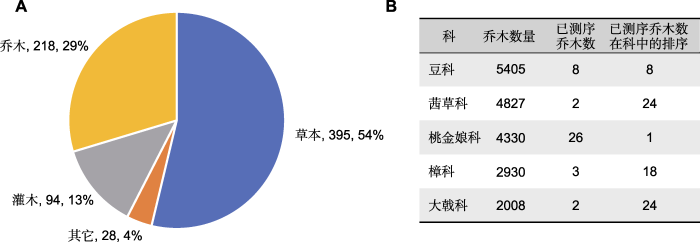
图1
已测序乔木物种概况
(A) 已测序物种的生活型分布, 图中依次标注生活型、数量和占已测序维管植物数量的百分比(附录1); (B) 乔木数量最多的5个科测序情况。已测序物种数据来自Sun等(2022b); 生活型数据来自GIFT数据库(Weigelt et al., 2020)
Figure 1
Overview of sequenced tree species
(A) The distribution of the growth form of sequenced species, growth form, species number and the percentage of the number of sequenced vascular plants were indicated (Appendix 1); (B) Overview of sequenced top five families with the largest number of tree species. The datasets of sequenced species were obtained from the paper of Sun et al. (2022b); the corresponding datasets of growth form were obtained from the GIFT database (Weigelt et al., 2020)
樟科是乔木物种数量排第4的大科(Beech et al., 2017), 全世界约有50属, 2 500-3 000种, 包含9个主要分支, 即樟族(Cinnamomeae)、月桂族(Laureae)、鳄梨族(Perseeae)、桂土楠群(Mezilaurus Group)、檬果樟族(Caryodaphnopsideae)、新樟族(Neocinnamomeae)、无根藤族(Cassytheae)、厚壳桂族(Cryptocaryeae)和棠桂族(Hypodaphnideae), 其中绝大多数为木本, 仅无根藤属(Cassytha) 20余种为草质寄生藤本(Rohwer, 1993; The Angiosperm Phylogeny Group, 2016)。樟科是湿润森林中常见的乔木类群, 也是亚热带常绿阔叶林的优势类群, 具有重要的生态价值(Rohwer, 1993; Liu et al., 2021)。樟科是原始被子植物花演化研究的重要类群, 隶属原始被子植物木兰分支樟目(Laurales), 具有未分化的原始花被片、花序类型和花性别变异(两性花和单性花) (The Angiosperm Phylogeny Group, 2016; Chen et al., 2020b)。樟科植物富含多种次生代谢产物, 具有特殊气味, 使得其成为重要的药品、香料和香水原料(中国科学院中国植物志编辑委员会, 1982; Chen et al., 2020b)。由于其包含特有次生代谢产物, 特别是多种芳香族化合物, 樟科植物适用于研究植物的化学成分、生态功能和进化关系(Chen et al., 2020a; Xiong et al., 2022)。从基因组水平探讨樟科的进化、花形态演化的分子证据及特殊气味的遗传基础, 不仅对揭示樟科植物的生物学特性、开发应用及遗传育种具有指导作用(Chen et al., 2020a), 也为阐明被子植物花器官演化、优良木材和芳香族化合物的形成机制提供新见解。
1 樟科全基因组研究进展
1.1 基因组研究概况
自2019年樟科第1个全基因组, 即牛樟(Cinnamomum kanehirae=Camphora kanahirae) (Chung and Hsieh, 2023)的基因组序列发表以来(Chaw et al., 2019), 樟科全基因组研究呈现蓬勃发展的态势。目前已有8个物种完成测序(图2; 表1), 分布于樟科3个族(3/9) 6个属(约6/50)。牛樟基因组不仅是樟科首个完成全基因组测序的物种, 也是木兰分支第1个测序的物种(Chaw et al., 2019)。该研究从全基因组层面探讨了木兰类与双子叶植物的关系, 发掘了与牛樟形成相关的基因。随后, 在2019-2020年, 樟科重要的水果鳄梨(Persea americana)、化学物质原料山苍子(Litsea cubeba)以及优良木材树种闽楠(Phoebe bournei)的基因组相继发表(Rendón-Anaya et al., 2019; Chen et al., 2020a, 2020b)。这些物种基因组丰富了樟科基因组数据库, 为樟科基因组研究奠定了基础。
图2
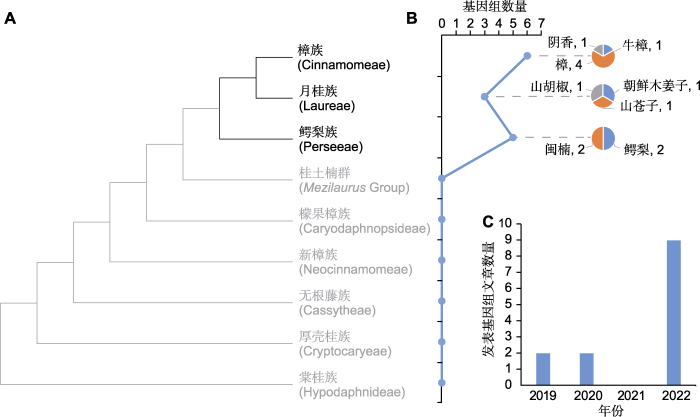
图2
樟科基因组已测序物种概况
(A) 樟科系统发生树(参考Liu et al., 2021); (B) 樟科物种已测序基因组数量折线图和各族测序物种基因组数量的饼图; (C) 近几年发表樟科基因组文章数量
Figure 2
Overview of sequenced species of Lauraceae
(A) Phylogenetic tree of Lauraceae (refer to Liu et al., 2021); (B) Line chart of the number of sequenced genome of Lauraceae, and pie charts of numbers of sequenced species of every tribe; (C) Number of articles on Lauraceae genome in recent years
表1 已测序樟科基因组信息
Table 1
| 序号 | 物种 | 族 | 基因组大小 (Mb) | 染色体 数目 (2n) | 杂合度 (%) | 编码基因数量 | 染色体挂载率 (%) | 组装完 整度 (BUSCO) (%) | 注释完 整度(BUSCO) (%) | 测序方法 | 组装 水平 | 参考文献 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 山苍子(Litsea cubeba) | 月桂族(Laureae) | 1325.7 | 24 | - | 31329 | 94.6 | 88.4 | 89.2 | PacBio CLR和Hi-C | 染色体 | Chen et al., 2020b |
| 2 | 朝鲜木姜子 (L. coreana) | 1139.5 | 24 | 1.1 | 32445 | 97.1 | 94.0 | - | Illumina、PacBio CCS和Hi-C | 染色体 | Zhang et al., 2022 | |
| 3 | 山胡椒(Lindera glauca) | 2092.2 | 24 | 1.4 | 65145 | 94.4 | 94.2 | 92.3 | Illumina、Nanopor和Hi-C | 染色体 | Xiong et al., 2022 | |
| 4 | 牛樟(Cinnamomum kanehirae=Camphora kanahirae) | 樟族(Cinnamomeae) | 730.7 | 24 | - | 27899 | - | 89.0 | - | Illumina、PacBio CLR、‘Chicago’和Hi-C | 染色体 | Chaw et al., 2019 |
| 5 | 樟(C. camphora=Ca. officinarum) | 755.4 | 24 | - | 24883 | 92.4 | 96.2 | - | PacBio CCS和Hi-C | 染色体 | Jiang et al., 2022 | |
| 6 | 723.1 | 24 | 1.2 | 36411 | 97.9 | 95.2 | 90.0 | Illumina、PacBio CCS和Hi-C | 染色体 | Wang et al., 2022 | ||
| 7 | 719.9 | 24 | 2.9 | 28789 | 99.9 | 95.3 | 89.8 | Illumina、PacBio CCS和Hi-C | 染色体 | Sun et al., 2022b | ||
| 8 | 785.0 | 24 | - | 29919 | 85.4 | 95.2 | 90.8 | PacBio CCS和Hi-C | 染色体 | Shen et al., 2022 | ||
| 9 | 阴香(C. burmanni) | 1177.6 | 24 | 0.7 | 41549 | 98.8 | 89.7 | - | Illumina、PacBio CLR和Hi-C | 染色体 | Li et al., 2022 | |
| 10 | 闽楠(Phoebe bournei) | 鳄梨族(Perseeae) | 989.2 | 24 | 1.5 | 28198 | - | 95.0 | 81.0 | PacBio CLR | Scaffold | Chen et al., 2020a |
| 11 | 941.8 | 24 | 1.4 | 30096 | 99.2 | 92.1 | 91.7 | PacBio CLR、BioNano和Hi-C | 染色体 | Han et al., 2022 | ||
| 12 | 鳄梨(Persea americana) | 912.6 | 24 | - | 24616 | 46.2 | 85.0 | - | PacBio CLR | 染色体 | Rendón-Anaya et al., 2019 | |
| 13 | 913.0 | 24 | - | 42769 | 98.8 | 98.9 | 96.6 | Illumina和PacBio CCS | 染色体 | Nath et al., 2022 |
2022年是樟科基因组研究成果爆发之年, 三代测序技术的发展特别是PacBio公司循环共识测序模式(circular consensus sequencing, CCS)生成HiFi reads (high fidelity reads)的技术促进了樟科全基因组测序研究(Sun et al., 2022a)。仅一年间共有9篇樟科基因组文章发表(图2C; 表1), 樟(Camphora officinarum) (Jiang et al., 2022; Shen et al., 2022; Sun et al., 2022a; Wang et al., 2022)、阴香(Cinnamomum burmanni) (Li et al., 2022)、朝鲜木姜子(Litsea coreana) (Zhang et al., 2022)及山胡椒(Lindera glauca) (Xiong et al., 2022)的全基因组序列相继发布, 鳄梨和闽楠的基因组质量得到提升(Han et al., 2022; Nath et al., 2022)。尤其值得关注的是, 樟作为我国重要的经济树种, 2022年有4个高质量基因组由不同单位主导发表(图2B; 表1)。
1.2 基因组特征
基因组大小及染色体数目是基因组的基本特征, 是研究科内多倍化和进化的基础。迄今为止, 樟科染色体数目的研究涵盖24个属(约24/50) 136个种(约136/ 3 000) (Oginuma and Tobe, 2006; Rice et al., 2015)。樟科物种多为二倍体, 通常n=12 (图3; 附录2)。染色体数目变化在樟科属内十分常见, 其中月桂属(Laurus)染色体n=18, 21, 24, 27, 30, 33, 36, 表现出丰富的变异。根据已发表的樟科基因组数据(包括全基因组测序的8个物种) (表1; 附录3), 樟科基因组大小为719 (樟)-2 982 Mb (月桂(Laurus nobilis))。月桂族的基因组明显大于樟族和鳄梨族(图4A)。
图3
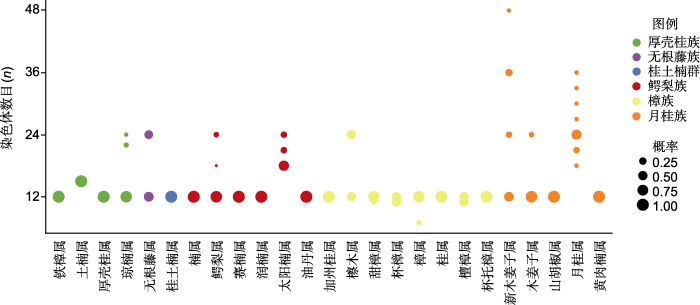
图3
樟科各属染色体数目变化
概率为染色体数目在该属中出现的比例。数据来自染色体数目数据库(CCDB, version 1.66) (Rice et al., 2015)及Oginuma和Tobe (2006) (附录2)
Figure 3
Variation in chromosome number of various genera in Lauraceae
Cirde sizes represent the percentage occurrence of chromosome numbers across genera. The data was extracted from chromosome counts database (CCDB, version 1.66) (Rice et al., 2015) and result from Oginuma and Tobe (2006) (Appendix 2)
图4
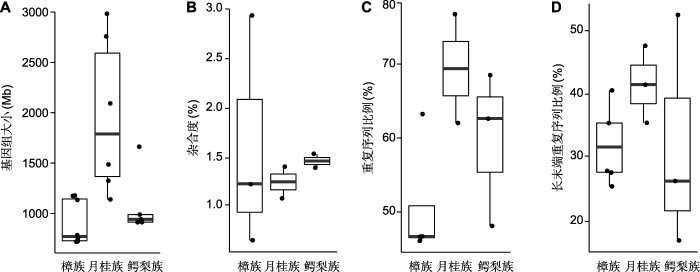
图4
已测序樟科基因组特征比较
(A) 基因组大小; (B) 基因组杂合度; (C) 重复序列比例; (D) 长末端重复序列比例
Figure 4
Comparison of characters of sequenced Lauraceae genomes
(A) Genome sizes; (B) Genomic heterozygosity; (C) Ratio of repeat sequences; (D) Ratio of long terminal repeat
基因组大小、杂合度和重复序列比例等是判断基因组复杂程度的标准(高胜寒等, 2018)。复杂基因组在测序和组装过程中会面临更大的困难(高胜寒等, 2018)。已测序樟科物种均为二倍体(2n=24)木本植物(表1), 基因组大小介于719-2 092 Mb之间。其中山胡椒基因组是已测序樟科物种中最大的基因组, 约为其它月桂族测序物种的2倍(表1)。樟科物种的基因组杂合度较高(图4B), 除阴香的杂合度为0.7%外, 其余物种的杂合度均大于1%。Sun等(2022a)测序的樟基因组杂合度高达2.9%, 是已测序樟科基因组中杂合度最高的。樟科基因组重复序列比例为46.1%-76.8%, 在樟科基因组中变化较大(图4C)。月桂族重复序列占比最高, 超过60%, 其中山胡椒的重复序列占比最高, 为76.8%。长末端重复序列(long terminal repeat, LTR)为樟科基因组中最常见的转座子(transposable elements, TE)。LTR在樟科已发表基因组中占比为17.0%- 52.5% (图4D), 在鳄梨基因组中比例最低, 但在闽楠基因组中比例最高(Chen et al., 2020a)。
1.3 起源和进化
1.3.1 木兰类的系统发生关系
通过基因组测序获取大量的单拷贝核基因数据并构建生命之树是探讨类群之间系统发生关系的重要方式(王伟和刘阳, 2020)。木兰类、真双子叶植物和单子叶植物之间的进化关系一直困扰着植物学家(Guo et al., 2023)。尽管已有许多木兰类基因组发表, 也利用转录组、叶绿体和线粒体数据构建了被子植物系统发生树(One Thousand Plant Transcriptomes Initiative, 2019; Li et al., 2021; Hu et al., 2023a), 但木兰类、真双子叶植物和单子叶植物之间的进化关系仍然存在争议(Shen et al., 2022)。樟科作为木兰类最大的科(The Angiosperm Phylogeny Group, 2016), 木兰类的系统发生位置也是樟科基因组研究的重点内容之一。樟科基因组相关文章中有11篇对该问题进行了讨论(占84.6%)。在樟科基因组研究结果中, 木兰类的系统发生位置呈现2种结构(图5B): I类是真双子叶植物的姐妹群, II类是真双子叶植物和单子叶植物构成分支的姐妹群。其中结构I更常见, 得到7篇文章的支持。
图5
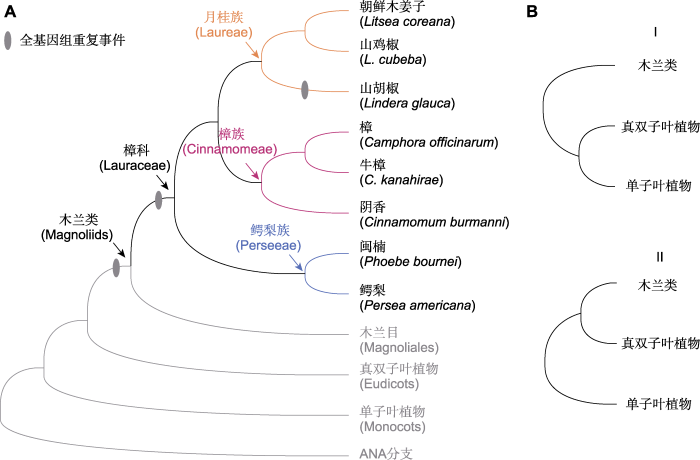
图5
樟科已测序物种系统发生关系
(A) 樟科已测序物种系统发生树(参考Liu et al., 2021; Han et al., 2022) (灰色椭圆形代表樟科经历的全基因组重复事件); (B) 从樟科基因组文献中得到的木兰类、真双子叶植物和单子叶植物系统发生关系的2种拓扑结构。
Figure 5
Phylogenetic relationships of sequenced species of Lauraceae
(A) Phylogenetic tree of sequenced species in Lauraceae (refer to Liu et al., 2021; Han et al., 2022) (The grey ovals represent predicted whole genome duplication events in Lauraceae); (B) Two topologies of the relationship of magnoliids, eudicots and monocots in the published papers based on the studies on Lauraceae genomes
在构建系统发生树时, 使用不同的采样策略、数据类型、基因数量和分析方法都会导致木兰类系统发生的结构不一致(Chen et al., 2020b; Han et al., 2022; Zhang et al., 2022; Hu et al., 2023a)。已发表的樟科基因组文献中, 研究者通常仅使用100-300个单拷贝直系同源基因构建系统发生树, 且取样缺少代表性。例如, 作为木兰类姐妹群的金粟兰目物种只包括在Wang等(2022)的基因组研究中。稀疏的分类单元取样和有限的基因数量会影响拓扑结构的可靠性, 而不同的数据类型和分析方法也会产生不同的结果。Chen等(2020a)基于核苷酸序列使用贝叶斯法构建的系统发生树以及基于氨基酸序列使用串联法和溯祖法构建的系统发生树均支持拓扑结构I, 而基于核苷酸序列使用串联法和溯祖法构建的系统发生树支持拓扑结构II。Zhang等(2022)基于核苷酸和氨基酸序列使用串联法构建的系统发生树支持拓扑结构I, 而用溯祖法所得结果支持拓扑结构II。不完全谱系分选(incomplete lineage sorting, ILS)被认为是造成木兰类系统发生关系不一致的重要因素(Rendón-Anaya et al., 2019; Chen et al., 2020a, 2020b; Zhang et al., 2022), 但仅有少量研究进行了相关分析。基因家族周转率分析(turnover analysis)表明, 在主要被子植物分支同时扩张的过程中存在明显的重复基因更替, 因此增加了发生不完全谱系分选的可能性(Rendón-Anaya et al., 2019)。基于溯祖法的ASTRAL分析表明, 早期被子植物快速分化时可能发生了不完全谱系分选(Chen et al., 2020a; Zhang et al., 2022)。除系统发生树外, 共线性分析可为木兰类的进化关系提供更多证据。在鳄梨基因组研究中, Rendón-Anaya等(2019)基于共线性的系统发生分析, 利用位于同源共线性模块上的同源基因序列差异, 支持木兰类为单子叶植物和双子叶植物的姐妹类群。
1.3.2 樟科的系统发生关系
自2000年樟科的首篇分子系统学文章发表以来(Rohwer, 2000), 科学家为探讨樟科物种的系统发生关系作出了许多努力, 但樟科的系统发生研究仍处于初级阶段(Tian et al., 2021)。究其原因主要有2点。(1) 取样代表性不足。Liu等(2021)利用叶绿体基因组构建了樟科目前取样最全且支持率最高的系统发生树, 但其取样仅占樟科属的50% (约25/50), 种的4% (约129/3 000)。形态性状变异小、形态的平行演化或逆转导致物种分类研究比较困难, 以及乔木生长在难以到达的热带森林中, 造成调查和采样困难, 是影响樟科取样代表性的2个主要因素(Li et al., 2004; 李捷和李锡文, 2004; Tian et al., 2021)。(2) 分子标记信息不足。常用的分子片段, 如matK、trnL-trnF、 trnT-trnL和psbA-trnH的分辨率不足以构建稳定的系统发生树(Rohwer, 2000; Chanderbali et al., 2001; Li et al., 2004, 2008; Rohwer and Rudolph, 2005), 而樟科ITS序列存在假基因和不完全一致性进化, 造成直接测序获得的序列有严重的杂合现象, 无法单独提供可靠的节点推断(黄建峰等, 2016)。虽然通过使用叶绿体基因组确定了樟科的基本框架, 但樟科叶绿体基因组十分保守, 无法解决各分支内属的系统发生关系(Xiao et al., 2020, 2022; Trofimov et al., 2022; Xiao and Ge, 2022)。
全基因组研究为樟科多层次系统发生提供了证据和大量的数据, 揭示了樟科复杂的进化历史。在山苍子基因组研究中, 研究人员结合樟科代表属的浅层基因组和转录组数据, 从基因组层面为樟科复杂的系统发育关系提供了新的证据(Chen et al., 2020b)。该研究从樟科转录组数据中鉴定出275个单拷贝核基因, 采用串联法和溯祖法推断樟科的系统发生关系, 构建了樟科19属46种的质体系统发生树。研究显示樟科核基因树与前人构建的质体树有显著差异(Liu et al., 2021), 且不一致的节点存在基因树和物种树的冲突。在质体树中, 厚壳桂属(Cryptocarya)是樟科其它类群的姐妹群, 而核基因树中樟科唯一的半寄生藤本类群无根藤属是其它类群的姐妹群(Chen et al., 2020b)。此结果与以往基于核基因和质体分子片段得到的不一致拓扑结构相似(Rohwer, 2000; Chanderbali et al., 2001; Rohwer and Rudolph, 2005)。在系统发生基因组学中, 不一致的进化历史推断十分普遍。不完全谱系分选、杂交(hybridization)和基因渐渗(gene introgression)等生物因素以及数据处理过程中的随机和系统误差等非生物因素都可能造成系统发生树不一致(王伟和刘阳, 2020; Steenwyk et al., 2023)。樟科系统发生树出现明显的拓扑结构冲突, 强调了杂交起源和网状进化对其类群系统发生关系的重要影响(Liu et al., 2021)。而月桂族、樟族和鳄梨族经历了快速辐射分化, 极有可能受到了不完全谱系分选的影响(Xiao et al., 2020, 2022; Liu et al., 2021; Xiao and Ge, 2022)。
在属级层面, 基因组数据也为确立广义樟属及其属下等级的系统发生关系提供了新见解。在樟的基因组研究方面, 首次使用了重测序方法探究广义樟属(Cinnamomum s.l.)内物种的亲缘关系(Wang et al., 2022)。利用重测序数据获得的单核苷酸多态性位点(single nucleotide polymorphisms sites, SNPs)和插入缺失位点(insertions and deletions sites, InDels)构建的广义樟属24个物种系统发生树显示, 樟属(Camphora)和桂属(Cinnamomum)植物形成两大分支。此外, 通过比较基因组学, Li等(2022)发现相较于牛樟和樟, 染色体结构和LTR的变化使阴香具有更为独立的进化历史。这些结果支持Yang等(2022a)将广义樟属划分为樟属和桂属的处理。
1.3.3 樟科的多倍化事件
樟科基因组研究中鉴定出3次多倍化事件(图2A)。在樟科所有测序基因组中都检测到木兰目和樟目分化之前发生的1次WGD事件(约118-147 Ma) (Sun et al., 2022a; Han et al., 2022)以及已测序樟科物种共同祖先中发生的1次WGD事件(约76-95 Ma) (Shen et al., 2022; Han et al., 2022)。较近的樟科WGD事件与晚白垩纪早期樟科植物随着冈瓦纳大陆的分裂快速辐射分化的时间一致(Chanderbali et al., 2001)。WGD事件通过改善基本生理活动和初级代谢使植物对环境变化的适应性增强, 促进了早期樟科物种的快速辐射(Chen et al., 2020a; Jiang et al., 2022; Sun et al., 2022a)。值得注意的是, 山胡椒基因组在与相近的山苍子分化后(14.90-23.18 Ma)独自经历了1次多倍化事件, 与早中新世(23 Ma)月桂族快速辐射分化的时间一致(Qin et al., 2023)。月桂族为樟科中雌雄异株类群, 主要分布在亚洲热带和亚热带地区, 为亚热带常绿阔叶林的建群种(Li et al., 2004; Qin et al., 2023)。这次多倍化事件导致山胡椒具有比山苍子及其它樟科植物大将近1倍的基因组和更多的蛋白质编码基因(表1) (Xiong et al., 2022)。其中, 与萜烯合酶生物合成相关的基因家族发生显著扩张, 可能对山胡椒的生态适应性和生物适应性有重要作用(Pichersky and Raguso, 2018; Xiong et al., 2022), 有利于其应对早中新世东亚季风盛行引起的极端天气(Qin et al., 2023)。
1.4 功能基因和基因家族
在樟科基因组中, 花器官进化、木材生长、代谢产物合成和炭疽病抗性等相关基因已得到研究(图6)。通过山苍子基因组结合转录组数据, 发现1个高度保守的参与花序形态发生的基因FUWA (PETAL LOSS)所构建的花序系统发育与樟科物种的系统进化相对应, 揭示了樟科花序从穗状花序、穗状圆锥花序向聚伞形圆锥花序、假伞形花序, 进而到伞形花序演变的规律(Chen et al., 2020b)。Sun等(2022a)在樟的基因组研究中通过花发育相关基因的表达分析和qRT- PCR验证早期被子植物花器官广泛活跃的祖先ABCDE模型。Nath等(2022)解析了鳄梨基因组中独特的庚糖生物合成途径, 发现其涉及景天庚酮糖1,7-二磷酸(sedoheptuloase 1,7-bisphosphate)这一中间体。在其基因组中还观察到大量内切葡聚糖酶基因, 支持鳄梨具有与纤维素酶有关的果实成熟机制, 这与大多数水果以内聚半乳糖醛酸酶作为糖苷酶和水解酶参与植物组织的浸渍以及软腐烂的机制不同(Wakabayashi, 2000)。Wang等(2022)通过比较樟和其它樟科植物鳄梨、牛樟、闽楠和山苍子的基因组, 筛选出中碳链油脂合成的关键候选基因FatB1 (终止脂肪酸碳链延伸)、type-B LPAAT和DGAT2b (参与甘油三酯装配), 并绘制出中链甘油三酯(medium-chain triglyceride)合成途径。
图6
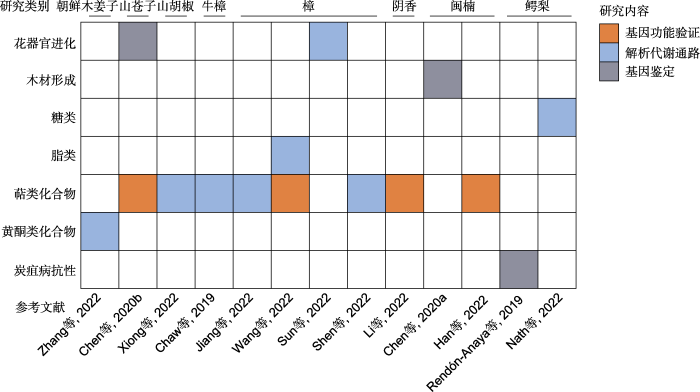
图6
樟科基因组功能基因研究热点
Figure 6
Research hotpots of functional genes of the Lauraceae genomes
樟科植物基因组中功能基因的鉴定涉及内容丰富, 但整体研究不够深入, 仍停留在基因鉴定和解析代谢通路层面, 仅有萜类化合物相关基因进行了功能验证(图6)。萜类化合物主要包括单萜、倍半萜和不规则萜类, 是结构多样化代谢产物中最大的一类(Christianson, 2017)。它们在植物生理功能中起至关重要的作用, 包括生长发育、环境适应、吸引传粉者以及抗病虫害和高温, 也是花和果实香味形成的重要活性物质(Nagegowda, 2020; Jia et al., 2022)。萜类合成酶(terpene synthase, TPS)是植物多样化萜类骨架形成的关键, 将甲基赤藓糖醇-4-磷酸途径(methylerythritol-4-phosphate pathway, MEP)或甲羟戊酸途径(mevalonate pathway, MVA)产物进一步催化生成各种萜烯类化合物(Jia et al., 2022)。樟科植物产生的挥发性有机化合物在香料和香水工业中具有很高的应用价值(中国科学院中国植物志编辑委员会, 1982)。不同樟科树种产生的芳香族化合物成分不同, 主要是萜类及其衍生物(Xiong et al., 2022), 山苍子、月桂和樟的气味主要由不同的单萜类化合物组成, 而楠属物种气味主要为倍半萜类化合物(Joshi et al., 2009; Han et al., 2022)。因此, 揭示萜类合酶基因家族重要基因的代谢途径, 探究萜类合酶基因家族在樟科中的演化是樟科基因组研究的热点。
基于基因组、转录组和代谢组等多组学数据的联合分析, 研究者鉴定出樟科中参与重要萜类化合物合成的关键基因并绘制了合成代谢通路图。通过转录组分析、同源和异源瞬时过表达以及体外酶活实验, 鉴定了调控山苍子主要单萜化合物合成的关键基因(Chen et al., 2020b)。其中LcuDXS3在果实中高表达, 可正调控单萜类和倍半萜类多种成分的含量; LcuTPS22在叶片中特异性积累, 调控樟科植物叶片中主要挥发性成分α-蒎烯、β-蒎烯、桉油醇、莰烯、桉油醇和樟脑的积累; LcuTPS42调控芳樟烯、香叶醇和樟科特定香味的主要成分芳樟醇的生物合成。山胡椒基因组研究揭示了其果实和叶片香气中的主要单一萜烯烃类化合物、β-罗勒烯与D-吉玛烯合成代谢过程中关键酶基因的变化规律, 绘制出合成代谢通路图(Xiong et al., 2022)。Wang等(2022)通过比较樟的5种化学型叶片转录组, 筛选差异表达的TPS基因, 并通过异源表达-体外催化反应与亚细胞定位等实验验证了CcTPS16和CcTPS54分别调控1,8-桉叶油素和异橙花叔醇的合成。Li等(2022)通过阴香的比较基因组学和2种化学型的转录组学及基因克隆和qPCR分析, 确定了右旋龙脑合成的2条关键分子途径及相关的功能基因, 推测MEP途径可能是阴香合成右旋龙脑的关键途径, 而CbTPS-1可能是导致阴香叶片右旋龙脑高产的关键调控基因之一。
串联重复、片段重复和WGD扩张的基因家族为进化提供了原始的遗传物质(Shen et al., 2022; Wang et al., 2022)。樟科物种中2次WGD事件可能造成其最近共同祖先中TPS基因大量扩张, 随后的串联重复显著促进了TPS基因家族的多样化, 特别是TPS-a和TPS-b亚家族(Chaw et al., 2019; Chen et al., 2020b; Han et al., 2022; Jiang et al., 2022; Li et al., 2022; Wang et al., 2022)。串联重复基因表现出较高的进化速率、较近的进化历史且在染色体上分布更聚集(Jiang et al., 2022)。在闽楠基因组中已经检测到TPS-a和TPS-b分支受到正向选择(Han et al., 2022), 从牛樟基因组中也检测到樟科特有的TPS-f-I和-II子分支受到正向选择(Chaw et al., 2019)。TPS基因亚家族之间不同的进化机制促进了萜类化合物在樟科的多样化。樟科祖先物种TPS基因大量扩张, 随后不同类群的基因经历了不同的扩张和收缩过程(Han et al., 2022)。牛樟和山苍子中单萜类萜烯合酶TPS-b亚家族成员较多, 而倍半萜类萜烯合酶TPS-a亚家族成员较少, 但闽楠基因组中TPS基因家族扩张后保留了更多的倍半萜类合成酶TPS-a亚家族成员, 丢失了大量的单萜合酶TPS-b拷贝(Han et al., 2022)。TPS基因亚家族在樟科各类群中分布的差异与物种的化学组分一致, 这也揭示了樟科各分支在适应性进化过程中经历了不同的进化历史。
2 研究展望
已往的樟科基因组研究探讨了被子植物木兰分支的系统发生关系和花器官进化, 重建了樟科系统发生树, 阐明了次生代谢产物合成的分子机制, 取得了重要进展, 为揭示樟科的进化历史和挖掘重要性状调控基因提供了基础信息。然而, 作为重要的林木类群, 樟科基因组研究仍有许多值得进一步探讨的问题。
总体来说, 樟科基因组研究呈现3个明显的特点。(1) 樟科基因组特征信息十分匮乏。除樟族、月桂族和鳄梨族以外的樟科物种基因组大小未见报道(图2; 表1)。(2) 已测序物种明显地向中国分布及有经济价值的物种倾斜。首先, 测序物种主要集中在樟和闽楠等作为化学物质原料或提供优质木材的经济树种。其次, 2019年以来一直是我国学者在主导樟科基因组研究, 已发表的基因组中, 仅鳄梨基因组由国外学者报道, 其余全部为中国学者对国产种类的研究报道。此外, 已发表的樟科基因组集中在核心樟类群(core Lauraceae), 对樟科原始分支棠桂族、厚壳桂族、新樟族、檬果樟族及无根藤族等缺少研究(图2)。最后, 在空间分布上, 测序种类表现出明显的亚洲分布倾向, 作为樟科分布中心的热带非洲和南美洲物种(除鳄梨外)未有涉及, 这也与参与樟科基因组研究的中国学者居多有关。(3) 功能基因的挖掘不够深入。目前, 学者们侧重于挖掘重要的功能基因并解析其分子机制, 但樟科基因组研究还停留在基因层面, 缺少功能基因的验证及具体机制探究, 除TPS基因家族在樟科的起源与进化得到探讨外(Han et al., 2022), 对功能基因的系统研究和比较分析十分缺乏(图6)。此外, 与花器官进化、木材形成和代谢产物合成相关的功能基因是研究热点, 而樟科作为被子植物基部类群, 其经历漫长的历史变迁仍在森林中占据优势的独特生物学特性尚未得到足够的关注(图6)。
综合目前的研究, 我们认为未来樟科基因组研究有以下4个值得深入的方向。
(1) 调查基因组的基本特征。染色体数目以及基因组大小等基本生物学信息是基因组研究的基础(Kress et al., 2022)。染色体数目对于染色体级别的全基因组组装极为重要, 但是樟科基因组的基础研究还十分缺乏(附录2, 附录3)。物种基因组数据能够帮助我们选择适合的测序物种, 优化测序和生物信息学研究方案, 以回答特定的生物学问题(Kress et al., 2022)。全基因组大小的多样性不仅在塑造植物基因组的进化中起重要作用, 而且在生态系统水平上影响植物群落的组合(Pellicer et al., 2018)。结合染色体数据研究基因组大小变化、染色体数量变化以及两者之间的相互作用机制有助于解释樟科多样化过程及基因组变化对其适应性进化的意义(Moeglein et al., 2020)。
(2) 增加测序基因组分支的代表性。目前樟科测序的基因组仅涉及3个族, 还有6个族尚无代表物种的基因组发表(图2; 表1)。樟科具有复杂的进化历史, 从系统发生拓扑结构不一致中可窥探一二。有代表性的物种取样能够帮助厘清樟科的系统发生关系。注释良好的基因组数据为使用重测序和靶向捕获的方法从标本中获取大量的基因序列提供参考, 这将解决樟科采集和鉴定难题, 从而构建更具有代表性的系统发生树(Sun et al., 2022a)。一方面, 解析樟科系统发生关系不仅可帮助我们从进化角度探讨特定功能基因的生物学特性和分子机制, 还能为保护濒危植物提供理论依据。桫椤(Alsophila spinulosa)基因组就是1个良好的案例(Huang et al., 2022)。研究者基于9个群体107个桫椤个体的基因组重测序解析树蕨类植物树干形成发育的独特性, 为理解蕨类植物的进化提供了新视角, 也从影响桫椤种群变化的因素中拓展了保护工作思路。另一方面, 基于系统发生树和分布数据可揭示樟科的时空分布格局, 解析其类群多样分布类型的成因。例如, 泛热带分布的厚壳桂属和琼楠属(Beilschmiedia) (Chanderbali et al., 2001), 东亚-北美间断分布的檫木属(Sassafras) (Yang et al., 2022b; Qin et al., 2023), 亚洲-大洋洲分布的土楠属(Endiandra) (Li et al., 2020), 以及仅分布在亚洲的新樟属(Neocinnamomun)和仅分布在美洲的桂土楠群(Chanderbali et al., 2001; Liu et al., 2021)。此外, 大量遗传信息和代表性取样能够为解析樟科复杂的进化历史提供支持(Liu et al., 2021; Tian et al., 2021)。目前, 樟科科内的多倍化只在山胡椒基因组中检测到(图5), 考虑到月桂族多样的染色体变化及其更大的基因组(图3), 增加月桂族物种全基因组测序, 有助于理解多倍化在樟科进化中的作用。
(3) 关注具有特殊价值的物种, 如月桂和无根藤(Cassytha filiformis)。月桂是唯一分布在欧洲且被赋予重要文化内涵的樟科植物, 其基因组具有复杂的染色体变化(附录2) (Chanderbali et al., 2001)。随着测序技术的进步及生物信息学软件的开发, 越来越多的复杂基因组得到解析, 如基因组大小为25.4 Gb、转座子含量达69.4%的油松(Niu et al., 2022); 已测序基金组最大的真双子叶植物杨山牡丹(Paeonia ostii), 其基因组大小为12.8 Gb (Yuan et al., 2022); 以及六倍体燕麦(Avena sativa) (Peng et al., 2022)。在新技术的推动下, 解析月桂基因组可探知樟科染色体结构的进化历史。而在原始被子植物樟科中, 无根藤寄生藤本的生活型一直备受科学家关注。解析无根藤的基因组将与已发表的寄生植物寄生花(Sapria himalayana) (Cai et al., 2021)和原野菟丝子(Cuscuta campestris) (Vogel et al., 2018)等共同为探究寄生植物的进化提供新视角。
(4) 对功能基因进行深入挖掘。樟科基因组功能基因的挖掘集中在与花器官进化、木材形成和代谢产物合成相关的基因, 且已挖掘的功能基因多数还未得到验证, 不利于基因的后续应用(图6)。解析与关键特征起源相关的功能基因和基因家族, 如落叶习性、单性花和寄生习性应受到更多关注。樟科因其特殊的系统发生位置, 在研究生物学特征的起源与进化上具有重要意义, 解析其关键特征起源的分子机制有助于揭开其作为原始类群至今繁盛的秘密。推动樟科重要物种高质量泛基因组研究将加深我们对其多样性的理解, 并发现与重要性状和环境适应相关的基因和变异, 为分子机制研究、品种选育和分子育种提供指导(Sun et al., 2022b; 郝晨路等, 2022)。以樟科重要的经济树种樟为例, 已发表的4个樟基因组在杂合度、编码基因数量和重复序列比例上具有明显差异(表1), 表现出个体间的遗传多样性(Jiang et al., 2022; Wang et al., 2022; Sun et al., 2022b; Shen et al., 2022)。泛基因组研究有助于我们理解种内个体间的差异, 进而更好地利用樟树资源。虽然目前泛基因组研究主要集中在农作物, 截至2021年末林木物种中仅苹果(Malus) (Wang et al., 2023b)和杨属(Populus) (Zhang et al., 2019)开展了泛基因组研究(郝晨路等, 2022)。但基因组规模大、复杂度高的林木物种泛基因组研究仍极具挑战性(Wang et al., 2023a)。随着技术的进步, 林木物种的泛基因组研究不仅能够促进对乔木起源的认识, 还能够解析森林建群种应对环境异质性的分子机制, 这对理解森林对环境变化的响应具有重要意义(郝晨路等, 2022; Wang et al., 2023a)。
作者贡献声明
杨智: 分析数据并撰写论文; 杨永: 技术支持并修改论文。
附录1 已测序维管植物科的生活型数量分布
Appendix 1 The distribution of the number of growth form in the families of sequenced vascular plants
附录2 樟科物种染色体数目信息
Appendix 2 Chromosome number of Lauraceae species
附录3 流式细胞仪测定的樟科基因组大小
Appendix 3 Genome size of Lauraceae estimated by flow cytometry
附录4 樟科TPS基因组亚家族数量
Appendix 4 Number of subfamilies in Lauraceae TPS genomes
参考文献

\n Amborella trichopoda\n is understood to be the most basal extant flowering plant and its genome is anticipated to provide insights into the evolution of plant life on Earth (see the Perspective by\n \n Adams\n \n ). To validate and assemble the sequence,\n \n Chamala\n et al.\n \n (p.\n 1516\n ) combined fluorescent in situ hybridization (FISH), genomic mapping, and next-generation sequencing. The\n Amborella Genome Project\n (p.\n 10.1126/science.1241089\n ) was able to infer that a whole-genome duplication event preceded the evolution of this ancestral angiosperm, and\n \n Rice\n et al.\n \n (p.\n 1468\n ) found that numerous genes in the mitochondrion were acquired by horizontal gene transfer from other plants, including almost four entire mitochondrial genomes from mosses and algae.\n
Lauraceae includes the genusPhoebe, and the family is linked to the evolution of magnoliids. We sequenced the genome ofPhoebe bourneiNanmu. The assembled genome size was 989.19 Mb, with a contig N50 value of 2.05 Mb. A total of 28,198 protein-coding genes were annotated inP. bournei. Whole-genome duplication (WGD) analysis showed that Lauraceae has experienced two WGD events; the older WGD event occurred just before the divergence of Lauraceae and Magnoliales, and the more recent WGD was shared by all lineages of Lauraceae. The phylogenetic tree showed that magnoliids form a sister clade to monocots and eudicots. We also identified 63 MADS-box genes, includingAGL12-like genes that may be related to the regulation ofP. bourneiroots andFIN219-like genes encoding GH3 proteins, which are involved in photomorphogenesis.SAUR50-like genes involved in light signal-mediated pedicel or stem development were also identified. FourATMYB46-and threePtrEPSP-homologous genes related to lignin biosynthesis were identified. These genes may be associated with the formation of straight trunks inP. bournei. Overall, theP. bourneireference genome provides insight into the origin, evolution, and diversification ofPhoebeand other magnoliids.
The laurel family within the Magnoliids has attracted attentions owing to its scents, variable inflorescences, and controversial phylogenetic position. Here, we present a chromosome-level assembly of theLitsea cubebagenome, together with low-coverage genomic and transcriptomic data for many other Lauraceae. Phylogenomic analyses show phylogenetic discordance at the position of Magnoliids, suggesting incomplete lineage sorting during the divergence of monocots, eudicots, and Magnoliids. An ancient whole-genome duplication (WGD) event occurred just before the divergence of Laurales and Magnoliales; subsequently, independent WGDs occurred almost simultaneously in the three Lauralean lineages. The phylogenetic relationships within Lauraceae correspond to the divergence of inflorescences, as evidenced by the phylogeny ofFUWA, a conserved gene involved in determining panicle architecture in Lauraceae. Monoterpene synthases responsible for production of specific volatile compounds in Lauraceae are functionally verified. Our work sheds light on the evolution of the Lauraceae, the genetic basis for floral evolution and specific scents.
Aims: Studies on plant systematics and evolution aim to elucidate the origin and diversification of plants and to elucidate the factors that affect the patterns of plant diversity temporally and spatially. As the largest and most highly attended conference on plant sciences, the International Botanical Congress (IBC) came to China in 2017 (IBC 2017) and attracted nearly 7,000 participants from around the globe. The success of holding IBC 2017 in China has profound influence on plant sciences in China both because it was an important forum to demonstrate the achievements and progresses of Chinese scientists and because it strengthened the link and collaboration of scientists between China and abroad. Progresses: On the occasion of the fifth anniversary of IBC 2017, I reviewed the achievements and breakthroughs in the field of plant systematics and evolution in China, including the origin and diversification of early plants, taxonomy and phylogenetic reconstruction of major plant lineages, plant speciation and adaptive evolution, species interaction and concerted evolution, origin and underlying mechanisms of innovation characters, plant polyploidy and polyploid evolution, species endangerment and protection, as well as origin and domestication of cultivated plants. All the progresses highlighted here have contributed greatly to our better understanding of plant biodiversity across the world, which embodies the growing impacts of Chinese scientists on global research and development in plant sciences. Prospect: I pointed out the opportunities and challenges that plant scientists have faced, including fast development of genome sequencing and ‘-omic’ studies, the interdisciplinary and multi-level investigations and cooperation, management of big data, and the practical applications of plant systematics and evolutionary studies in resource utilization, species conservation, agriculture and horticulture, medicine and plant trade, etc.
植物系统和进化生物学旨在探讨植物物种多样性的起源、多样化及其进化的机制, 是综合性越来越强的研究领域。2017年在深圳召开的第19届国际植物学大会(IBC 2017)为中国学者提供了一次难得的展示自身实力的机会和舞台, 同时也极大地推动了中国植物系统与进化生物学领域的研究。值此大会召开5周年之际, 本文拟就中国系统和进化生物学领域近年来取得的主要进展和突破做一简要回顾, 以帮助读者了解中国植物系统和进化研究的发展态势, 并在此基础上展望未来该领域的发展趋势以及面临的机遇和挑战。在过去5年中, 中国学者在植物系统与进化生物学领域的各个方面均取得了令人鼓舞的成绩和突破, 涉及植物起源和物种多样性格局的演变、植物分类和系统发生重建、物种形成和适应性进化、种间互作和协同进化、新性状的起源及其进化发育机制、植物多倍化的机制和多倍体进化、物种濒危机制和物种保护以及栽培植物的起源和驯化等等。这些研究成果不仅在数量上而且在质量上有显著提升, 受到国际学界的广泛关注, 意味着中国学者已经成为国际该领域研究的重要力量, 并将在国际植物系统和进化研究领域发挥更大的作用。
The advances accelerated by next-generation sequencing and long-read sequencing technologies continue to provide an impetus for plant phylogenetic study. In the past decade, a large number of phylogenetic studies adopting hundreds to thousands of genes across a wealth of clades have emerged and ushered plant phylogenetics and evolution into a new era. In the meantime, a roadmap for researchers when making decisions across different approaches for their phylogenomic research design is imminent. This review focuses on the utility of genomic data (from organelle genomes, to both reduced representation sequencing and whole-genome sequencing) in phylogenetic and evolutionary investigations, describes the baseline methodology of experimental and analytical procedures, and summarizes recent progress in flowering plant phylogenomics at the ordinal, familial, tribal, and lower levels. We also discuss the challenges, such as the adverse impact on orthology inference and phylogenetic reconstruction raised from systematic errors, and underlying biological factors, such as whole-genome duplication, hybridization/introgression, and incomplete lineage sorting, together suggesting that a bifurcating tree may not be the best model for the tree of life. Finally, we discuss promising avenues for future plant phylogenomic studies.
对樟科樟属(Cinnamomum Schaeffer) 17个代表样本的核糖体DNA内转录间隔区(nrDNA ITS)进行克隆测序。对获得的87条不同ITS序列的长度变异、GC含量、5.8S区二级结构的稳定性、遗传距离、进化模式以及系统发育关系进行了相关分析。研究结果显示, ITS序列在樟属植物内存在明显的多态性, 87条序列中的22条序列被鉴定为假基因序列, 其余65条序列为功能基因序列; 假基因序列采用中性进化模式, 变异明显大于功能序列。ITS序列在樟属植物中出现一致性进化不完全和假基因现象也可能发生在樟科其它类群中, 这可能是导致樟科植物ITS序列直接测序方式成功率低的重要原因。
\n Land plants produce numerous terpenoids that regulate development and mediate environmental interactions. Thus, how typical plant terpene synthase (\n TPS\n ) genes originated and evolved to create terpenoid diversity is of fundamental interest. By investigating\n TPSs\n from the genomes and transcriptomes of diverse taxa of green plants, it was demonstrated here that the ancestral\n TPS\n gene originated in land plants after divergence from green algae and encoded a bifunctional\n ent\n -kaurene synthase for phytohormone biosynthesis. This ancestral\n TPS\n then underwent gene duplication at least twice early in land plant evolution, leading to three ancient\n TPS\n lineages reflecting sub-functionalization of class I and II activities for phytohormone biosynthesis and neo-functionalization from primary to secondary metabolism, followed in each case by dynamic functional divergence.\n
Camphor tree [Cinnamomum camphora (L.) J. Presl], a species in the magnoliid family Lauraceae, is known for its rich volatile oils and is used as a medical cardiotonic and as a scent in many perfumed hygiene products. Here, we present a high-quality chromosome-scale genome of C. camphora with a scaffold N50 of 64.34 Mb and an assembled genome size of 755.41 Mb. Phylogenetic inference revealed that the magnoliids are a sister group to the clade of eudicots and monocots. Comparative genomic analyses identified two rounds of ancient whole-genome duplication (WGD). Tandem duplicated genes exhibited a higher evolutionary rate, a more recent evolutionary history and a more clustered distribution on chromosomes, contributing to the production of secondary metabolites, especially monoterpenes and sesquiterpenes, which are the principal essential oil components. Three-dimensional analyses of the volatile metabolites, gene expression and climate data of samples with the same genotype grown in different locations showed that low temperature and low precipitation during the cold season modulate the expression of genes in the terpenoid biosynthesis pathways, especially TPS genes, which facilitates the accumulation of volatile compounds. Our study lays a theoretical foundation for policy-making regarding the agroforestry applications of camphor tree.
Green plants play a fundamental role in ecosystems, human health, and agriculture. As de novo genomes are being generated for all known eukaryotic species as advocated by the Earth BioGenome Project, increasing genomic information on green land plants is essential. However, setting standards for the generation and storage of the complex set of genomes that characterize the green lineage of life is a major challenge for plant scientists. Such standards will need to accommodate the immense variation in green plant genome size, transposable element content, and structural complexity while enabling research into the molecular and evolutionary processes that have resulted in this enormous genomic variation. Here we provide an overview and assessment of the current state of knowledge of green plant genomes. To date fewer than 300 complete chromosome-scale genome assemblies representing fewer than 900 species have been generated across the estimated 450,000 to 500,000 species in the green plant clade. These genomes range in size from 12 Mb to 27.6 Gb and are biased toward agricultural crops with large branches of the green tree of life untouched by genomic-scale sequencing. Locating suitable tissue samples of most species of plants, especially those taxa from extreme environments, remains one of the biggest hurdles to increasing our genomic inventory. Furthermore, the annotation of plant genomes is at present undergoing intensive improvement. It is our hope that this fresh overview will help in the development of genomic quality standards for a cohesive and meaningful synthesis of green plant genomes as we scale up for the future.
Flowering plants (angiosperms) are dominant components of global terrestrial ecosystems, but phylogenetic relationships at the familial level and above remain only partially resolved, greatly impeding our full understanding of their evolution and early diversification. The plastome, typically mapped as a circular genome, has been the most important molecular data source for plant phylogeny reconstruction for decades.
Using DNA barcoding for species identification remains challenging for many plant groups. New sequencing approaches such as complete plastid genome sequencing may provide some increased power and practical benefits for species identification beyond standard plant DNA barcodes. We undertook a case study comparing standard DNA barcoding to plastid genome sequencing for species discrimination in the ecologically and economically important family Lauraceae, using 191 plastid genomes for 131 species from 25 genera, representing the largest plastome data set for Lauraceae to date. We found that the plastome sequences were useful in correcting some identification errors and for finding new and cryptic species. However, plastome data overall were only able to discriminate c. 60% of the species in our sample, with this representing a modest improvement from 40 to 50% discrimination success with the standard plant DNA barcodes. Beyond species discrimination, the plastid genome sequences revealed complex relationships in the family, with 12/25 genera being non-monophyletic and with extensive incongruence relative to nuclear ribosomal DNA. These results highlight that although useful for improving phylogenetic resolution in the family and providing some species-level insights, plastome sequences only partially improve species discrimination, and this reinforces the need for large-scale nuclear data to improve discrimination among closely related species.
Avocado (Persea americana) is a member of the magnoliids, an early branching lineage of angiosperms that has high value globally with the fruit being highly nutritious. Here, we report a chromosome-level genome assembly for the commercial avocado cultivar Hass, which represents 80% of the world’s avocado consumption. The DNA contigs produced from Pacific Biosciences HiFi reads were further assembled using a previously published version of the genome supported by a genetic map. The total assembly was 913 Mb with a contig N50 of 84 Mb. Contigs assigned to the 12 chromosomes represented 874 Mb and covered 98.8% of benchmarked single-copy genes from embryophytes. Annotation of protein coding sequences identified 48 915 avocado genes of which 39 207 could be ascribed functions. The genome contained 62.6% repeat elements. Specific biosynthetic pathways of interest in the genome were investigated. The analysis suggested that the predominant pathway of heptose biosynthesis in avocado may be through sedoheptulose 1,7 bisphosphate rather than via alternative routes. Endoglucanase genes were high in number, consistent with avocado using cellulase for fruit ripening. The avocado genome appeared to have a limited number of translocations between homeologous chromosomes, despite having undergone multiple genome duplication events. Proteome clustering with related species permitted identification of genes unique to avocado and other members of the Lauraceae family, as well as genes unique to species diverged near or prior to the divergence of monocots and eudicots. This genome provides a tool to support future advances in the development of elite avocado varieties with higher yields and fruit quality.
Genome size is a biodiversity trait that shows staggering diversity across eukaryotes, varying over 64,000-fold. Of all major taxonomic groups, land plants stand out due to their staggering genome size diversity, ranging ca. 2400-fold. As our understanding of the implications and significance of this remarkable genome size diversity in land plants grows, it is becoming increasingly evident that this trait plays not only an important role in shaping the evolution of plant genomes, but also in influencing plant community assemblages at the ecosystem level. Recent advances and improvements in novel sequencing technologies, as well as analytical tools, make it possible to gain critical insights into the genomic and epigenetic mechanisms underpinning genome size changes. In this review we provide an overview of our current understanding of genome size diversity across the different land plant groups, its implications on the biology of the genome and what future directions need to be addressed to fill key knowledge gaps.
Common oat (Avena sativa) is an important cereal crop serving as a valuable source of forage and human food. Although reference genomes of many important crops have been generated, such work in oat has lagged behind, primarily owing to its large, repeat-rich polyploid genome. Here, using Oxford Nanopore ultralong sequencing and Hi-C technologies, we have generated a reference-quality genome assembly of hulless common oat, comprising 21 pseudomolecules with a total length of 10.76 Gb and contig N50 of 75.27 Mb. We also produced genome assemblies for diploid and tetraploid Avena ancestors, which enabled the identification of oat subgenomes and provided insights into oat chromosomal evolution. The origin of hexaploid oat is inferred from whole-genome sequencing, chloroplast genomes and transcriptome assemblies of different Avena species. These findings and the high-quality reference genomes presented here will facilitate the full use of crop genetic resources to accelerate oat improvement.
All plants synthesize a suite of several hundred terpenoid compounds with roles that include phytohormones, protein modification reagents, anti-oxidants, and more. Different plant lineages also synthesize hundreds of distinct terpenoids, with the total number of such specialized plant terpenoids estimated in the scores of thousands. Phylogenetically restricted terpenoids are implicated in defense or in the attraction of beneficial organisms. A popular hypothesis is that the ability of plants to synthesize new compounds arose incrementally by selection when, as a result of gradual changes in their biotic partners and enemies, the 'old' plant compounds were no longer effective, a process dubbed the 'coevolutionary arms race'. Another hypothesis posits that often the sheer diversity of such compounds provides benefits that a single compound cannot. In this article, we review the unique features of the biosynthetic apparatus of terpenes in plants that facilitate the production of large numbers of distinct terpenoids in each species and how facile genetic and biochemical changes can lead to the further diversification of terpenoids. We then discuss evidence relating to the hypotheses that given ecological functions may be enhanced by the presence of mixtures of terpenes and that the acquisition of new functions by terpenoids may favor their retention once the original functions are lost.© 2016 The Authors. New Phytologist © 2016 New Phytologist Trust.
The genus Ocotea (Lauraceae) includes about 450 species, of which about 90% are Neotropical, while the rest is from Macaronesia, Africa and Madagascar. In this study we present the first complete chloroplast genome sequences of seven Ocotea species, six Neotropical and one from Macaronesia. Genome sizes range from 152,630 (O. porosa) to 152,685 bp (O. aciphylla). All seven plastomes contain a total of 131 (114 unique) genes, among which 87 (80 unique) encode proteins. The order of genes (if present) is the same in all Lauraceae examined so far. Two hypervariable loci were found in the LSC region (psbA-trnH, ycf2), three in the SSC region (ycf1, ndhH, trnL(UAG)-ndhF). The pairwise cp genomic alignment between the taxa showed that the LSC and SSC regions are more variable compared to the IR regions. The protein coding regions comprise 25,503-25,520 codons in the Ocotea plastomes examined. The most frequent amino acids encoded in the plastomes were leucine, isoleucine, and serine. SSRs were found to be more frequent in the two dioecious Neotropical Ocotea species than in the four bisexual species and the gynodioecious species examined (87 vs. 75-84 SSRs). A preliminary phylogenetic analysis based on 69 complete plastomes of Lauraceae species shows the seven Ocotea species as sister group to Cinnamomum sensu lato. Sequence divergence among the Ocotea species appears to be much lower than among species of the most closely related, likewise species-rich genera Cinnamomum, Lindera and Litsea.© 2022. The Author(s).
A parasitic lifestyle, where plants procure some or all of their nutrients from other living plants, has evolved independently in many dicotyledonous plant families and is a major threat for agriculture globally. Nevertheless, no genome sequence of a parasitic plant has been reported to date. Here we describe the genome sequence of the parasitic field dodder, Cuscuta campestris. The genome contains signatures of a fairly recent whole-genome duplication and lacks genes for pathways superfluous to a parasitic lifestyle. Specifically, genes needed for high photosynthetic activity are lost, explaining the low photosynthesis rates displayed by the parasite. Moreover, several genes involved in nutrient uptake processes from the soil are lost. On the other hand, evidence for horizontal gene transfer by way of genomic DNA integration from the parasite's hosts is found. We conclude that the parasitic lifestyle has left characteristic footprints in the C. campestris genome.
The gymnosperm Welwitschia mirabilis belongs to the ancient, enigmatic gnetophyte lineage. It is a unique desert plant with extreme longevity and two ever-elongating leaves. We present a chromosome-level assembly of its genome (6.8 Gb/1 C) together with methylome and transcriptome data to explore its astonishing biology. We also present a refined, high-quality assembly of Gnetum montanum to enhance our understanding of gnetophyte genome evolution. The Welwitschia genome has been shaped by a lineage-specific ancient, whole genome duplication (~86 million years ago) and more recently (1-2 million years) by bursts of retrotransposon activity. High levels of cytosine methylation (particularly at CHH motifs) are associated with retrotransposons, whilst long-term deamination has resulted in an exceptionally GC-poor genome. Changes in copy number and/or expression of gene families and transcription factors (e.g. R2R3MYB, SAUR) controlling cell growth, differentiation and metabolism underpin the plant’s longevity and tolerance to temperature, nutrient and water stress.
Due to the development of sequencing technology and the great reduction in sequencing costs, an increasing number of plant genomes have been assembled, and numerous genomes have revealed large amounts of variations. However, a single reference genome does not allow the exploration of species diversity, and therefore the concept of pan-genome was developed. A pan-genome is a collection of all sequences available for a species, including a large number of consensus sequences, large structural variations, and small variations including single nucleotide polymorphisms and insertions/deletions. A simple linear pan-genome does not allow these structural variations to be intuitively characterized, so graph-based pan-genomes have been developed. These pan-genomes store sequence and structural variation information in the form of nodes and paths to store and display species variation information in a more intuitive manner. The key role of graph-based pan-genomes is to expand the coordinate system of the linear reference genome to accommodate more regions of genetic diversity. Here, we review the origin and development of graph-based pan-genomes, explore their application in plant research, and further highlight the application of graph-based pan-genomes for future plant breeding.
Structural variations (SVs) and copy number variations (CNVs) contribute to trait variations in fleshy-fruited species. Here, we assemble 10 genomes of genetically diverse Malus accessions, including the ever-green cultivar ‘Granny Smith’ and the widely cultivated cultivar ‘Red Fuji’. Combining with three previously reported genomes, we assemble the pan-genome of Malus species and identify 20,220 CNVs and 317,393 SVs. We also observe CNVs that are positively correlated with expression levels of the genes they are associated with. Furthermore, we show that the noncoding RNA generated from a 209 bp insertion in the intron of mitogen-activated protein kinase homology encoding gene, MMK2, regulates the gene expression and affects fruit coloration. Moreover, we identify overlapping SVs associated with fruit quality and biotic resistance. This pan-genome uncovers possible contributions of CNVs to gene expression and highlights the role of SVs in apple domestication and economically important traits.
In 1859, Charles Darwin put forward the concept of the tree of life (TOL), a metaphor for charting relationships between organisms in space and time in his The Origin of Species. The TOL is a cornerstone in evolutionary theory and makes sense of all biology. Decades of research in plant molecular systematics has led to substantial progress in understanding many aspects of the plant TOL. Here, we summarized five major aspects of reconstructing the plant TOL, which are being studied at the present day and will continue to be goals moving forward. These include: (1) constructing genus- and species-level phylogenies for plant groups; (2) resolving deep-time and/or rapidly divergent phylogenetic relationships using genomic approaches; (3) updating classification systems by combining morphological and molecular data; (4) integrating fossil taxa into phylogenies derived from extant taxa; and (5) building big trees using supermatrix methods. We then outlined the current state of plant molecular systematics and highlight existing problems in the field, specifically in regard to China. Finally, we propose the corresponding guidelines and policy suggestions for the continued study of China’s reconstruction of the plant TOL.
Cinnamomum species attract attentions owing to their scents, medicinal properties, and ambiguous relationship in the phylogenetic tree. Here, we report a high-quality genome assembly of Cinnamomum camphora, based on which two whole-genome duplication (WGD) events were detected in the C. camphora genome: one was shared with Magnoliales, and the other was unique to Lauraceae. Phylogenetic analyses illustrated that Lauraceae species formed a compact sister clade to the eudicots. We then performed whole-genome resequencing on 24 Cinnamomum species native to China, and the results showed that the topology of Cinnamomum species was not entirely consistent with morphological classification. The rise and molecular basis of chemodiversity in Cinnamomum were also fascinating issues. In this study, six chemotypes were classified and six main terpenoids were identified as major contributors of chemodiversity in C. camphora by the principal component analysis. Through in vitro assays and subcellular localization analyses, we identified two key terpene synthase (TPS) genes (CcTPS16 and CcTPS54), the products of which were characterized to catalyze the biosynthesis of two uppermost volatiles (i.e. 1,8-cineole and (iso)nerolidol), respectively, and meditate the generation of two chemotypes by transcriptional regulation and compartmentalization. Additionally, the pathway of medium-chain triglyceride (MCT) biosynthesis in Lauraceae was investigated for the first time. Synteny analysis suggested that the divergent synthesis of MCT and long-chain triglyceride (LCT) in Lauraceae kernels was probably controlled by specific medium-chain fatty acyl-ACP thioesterase (FatB), type-B lysophosphatidic acid acyltransferase (type-B LPAAT), and diacylglycerol acyltransferase 2b (DGAT 2b) isoforms during co-evolution with retentions or deletions in the genome.
Ancient whole-genome duplications (WGDs or polyploidy) are prevalent in plants, and some WGDs occurred during the timing of severe global environmental changes. It has been suggested that WGDs may have contributed to plant adaptation. However, this still lacks empirical evidence at the genetic level to support the hypothesis. Here, we investigated the survivors of gene duplicates from multiple ancient WGD events on the major branches of angiosperm phylogeny, and aimed to explore genetic evidence supporting the significance of polyploidy. Duplicated genes co-retained from three waves of independent WGDs (∼120 million years ago [Ma], ∼66, and <20 Ma) were investigated in 25 selected species. Gene families functioning in low temperature and darkness were commonly retained gene duplicates after the eight independently occurring WGDs in many lineages around the Cretaceous-Paleocene boundary, when the global cooling and darkness were the two main stresses. Moreover, the commonly retained duplicates could be key factors which may have contributed to the robustness of the critical stress-related pathways. In addition, genome-wide transcription factors (TFs) functioning in stresses tend to retain duplicates after waves of WGDs, and the coselected gene duplicates in many lineages may play critical roles during severe environmental stresses. Collectively, these results shed new light on the significant contribution of paleopolyploidy to plant adaptation during global environmental changes in the evolutionary history of angiosperms.Copyright © 2019 The Author. Published by Elsevier Inc. All rights reserved.
Tribe Cinnamomeae is a species-rich and ecologically important group in tropical and subtropical forests. Previous studies explored its phylogenetic relationships and historical biogeography using limited loci, which might result in biased molecular dating due to insufficient parsimony-informative sites. Thus, 15 plastomes were newly sequenced and combined with published plastomes to study plastome structural variations, gene evolution, phylogenetic relationships, and divergence times of this tribe.
Gene tree discordance is common in phylogenetic analyses. Many phylogenetic studies have excluded non-coding regions of the plastome without evaluating their impact on tree topology. In general, plastid loci have often been treated as a single unit, and tree discordance among these loci has seldom been examined. Using samples of Laureae (Lauraceae) plastomes, we explored plastome variation among the tribe, examined the influence of non-coding regions on tree topology, and quantified intra-plastome conflict.
The East Asian subtropical evergreen broad-leaved forests (EBLFs) harbor remarkable biodiversity. However, their historical assembly remains unclear. To gain new insights into the assembly of this biome, we generated a molecular phylogeny of one of its essential plant groups, the tribe Perseeae (Lauraceae).
Sassafrashas been considered to belong to trib. Laureae of Lauraceae and has been assumed to have unisexual flowers. However, recent molecular phylogenetic studies have consistently suggested thatSassafrasdoes not belong to the trib. Laureae but to Cinnamomeae and that it is nested withinCinnamomum. A recent morphological study revealed that one of the Asian species,S. randaiense, possesses bisexual flowers that are plesiomorphic in the family Lauraceae. As reports on the flower structure of the second Asian species,S. tzumu, have been contradictory, we wanted to ascertain if it has bisexual flowers or not. If the flowers were bisexual, could earlier reports that they were unisexual have been based on dichogamous flowering?
Tree peony (Paeonia ostii) is an economically important ornamental plant native to China. It is also notable for its seed oil, which is abundant in unsaturated fatty acids such as α-linolenic acid (ALA). Here, we report chromosome-level genome assembly (12.28 Gb) of P. ostii. In contrast to monocots with giant genomes, tree peony does not appear to have undergone lineage-specific whole-genome duplication. Instead, explosive LTR expansion in the intergenic regions within a short period (~ two million years) may have contributed to the formation of its giga-genome. In addition, expansion of five types of histone encoding genes may have helped maintain the giga-chromosomes. Further, we conduct genome-wide association studies (GWAS) on 448 accessions and show expansion and high expression of several genes in the key nodes of fatty acid biosynthetic pathway, including SAD, FAD2 and FAD3, may function in high level of ALAs synthesis in tree peony seeds. Moreover, by comparing with cultivated tree peony (P. suffruticosa), we show that ectopic expression of class A gene AP1 and reduced expression of class C gene AG may contribute to the formation of petaloid stamens. Genomic resources reported in this study will be valuable for studying chromosome/genome evolution and tree peony breeding.© 2022. The Author(s).
The genus Populus comprises a complex amalgam of ancient and modern species that has become a prime model for evolutionary and taxonomic studies. Here we sequenced the genomes of 10 species from five sections of the genus Populus, identified 71 million genomic variations, and observed new correlations between the single-nucleotide polymorphism-structural variation (SNP-SV) density and indel-SV density to complement the SNP-indel density correlation reported in mammals. Disease resistance genes (R genes) with heterozygous loss-of-function (LOF) were significantly enriched in the 10 species, which increased the diversity of poplar R genes during evolution. Heterozygous LOF mutations in the self-incompatibility genes were closely related to the self-fertilization of poplar, suggestive of genomic control of self-fertilization in dioecious plants. The phylogenetic genome-wide SNPs tree also showed possible ancient hybridization among species in sections Tacamahaca, Aigeiros, and Leucoides. The pangenome resource also provided information for poplar genetics and breeding.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}