
 首页
首页

多倍化(polyploid)或全基因组复制(whole-genome duplication, WGD)是物种多样性发生的重要驱动力(De Bodt et al., 2005; Van de Peer et al., 2017; Mandáková and Lysak, 2018), 在植物演化历史中普遍存在, 尤其是维管束植物中多样性最高的类群被子植物和第二大类群蕨类反复发生过多轮全基因组复制(One Thousand Plant Transcriptomes Initiative, 2019; 汪浩等, 2019; Huang et al., 2020; 王婷等, 2021)。基于现有证据, 在蕨类植物、被子植物第一大科菊科(Asteraceae)、第三大科豆科(Fabaceae)中分别检测到19、41、28次全基因组复制事件(Huang et al., 2020; Zhang et al., 2021a; Zhao et al., 2021), 推测多倍化与蕨类植物和被子植物物种多样性较高类群的物种形成和多样化有关(De Bodt et al., 2005; Van de Peer et al., 2017; Mandáková and Lysak, 2018; Ren et al., 2018)。
全基因组复制使得染色体和基因组内全部基因均发生加倍, 为新性状演化和物种多样化提供了遗传材料(De Bodt et al., 2005; Wu et al., 2020)。而全基因组复制后的基因丢失、沉默、亚功能化和新功能化等基因水平的变异, 以及染色体重组等染色体水平变异促进了表型和物种的多样化(Wendel, 2000; Adams and Wendel, 2005; Mandáková and Lysak, 2018)。此外, 全基因组复制及后续变异导致一些类群染色体数目变异(Mandáková and Lysak, 2018)。以单子叶植物禾本科为例, 禾本科祖先的染色体基数为7条, 而在经历了全基因组复制事件后(Paterson et al., 2004; Salse et al., 2008), 水稻(Oryza sativa)、高粱(Sorghum bicolor)和谷子(Setaria italica)的染色体基数并未达到加倍后的14条, 而是表现为染色体数目不同程度地减少, 分别为12、10和9条(Murat et al., 2017; 王振怡和王希胤, 2020)。因此, 染色体数目的变化是全基因组复制发生及后续演化进程的重要特征之一。
兰科(Orchidaceae)含700余属、约26 000种, 为被子植物第二大科, 单子叶植物第一大科, 是陆生植物中极具多样性的类群之一(Chase et al., 2015; Li et al., 2016), 同时表现出染色体数目变化较大(染色体基数从x=6到x=120)的特点(Da Conceição et al., 2006; 王筠竹等, 2019), 表明兰科植物的演化过程可能存在多次全基因组复制事件。然而, 目前在兰科植物中已见报道的全基因组复制事件非常有限。基于兰科植物基因组证据(Cai et al., 2015; Zhang et al., 2016, 2017; Yuan et al., 2018; Hasing et al., 2020)以及千种植物转录组项目等转录组分析(One Thousand Plant Transcriptomes Initiative, 2019), 目前仅检测到1次兰科植物特异发生的全基因组复制事件, 与蕨类(Huang et al., 2020)、菊科(Zhang et al., 2021a)和豆科(Zhao et al., 2021)等物种多样性丰富的类群多倍化研究结果不符。
分析上述情况的原因, 我们推测可能与兰科植物种类及类群众多、前期研究样本量小但种类跨度大的研究策略有关。例如, 千种植物转录组项目囊括了兰科7个样本, 但却跨了香荚兰亚科(Vanilloideae)、兰亚科(Orchidoideae)和树兰亚科(Epidendroideae) 3个亚科7个属(One Thousand Plant Transcriptomes Initiative, 2019); 分析全基因组复制事件的5套全基因组数据同样覆盖了拟兰亚科(Apostasioideae)、香荚兰亚科、树兰亚科3个亚科5个属(Cai et al., 2015; Zhang et al., 2016, 2017; Yuan et al., 2018; Hasing et al., 2020); 关于杓兰亚科基因组进化的研究包括13个兰科植物转录组和基因组数据, 覆盖了兰科所有亚科(拟兰亚科、香荚兰亚科、杓兰亚科(Cypripedioideae)、兰亚科和树兰亚科) 13个属(Unruh et al., 2018)。对于兰科这样包含26 000多种的特大类群, 解析其全基因组复制历史需要借助更精细的尺度。
杓兰亚科具有囊状或倒盔状唇瓣、2个可育雄蕊和1个盾状退化雄蕊等特征, 是兰科多样性的重要代表类群之一, 包括杓兰属(Cypripedium)、南美杓兰属(Selenipedium)、美洲兜兰属(Phragmipedium)、镊萼兜兰属(Mexipedium)及兜兰属(Paphiopedilum) 5个属(Cox et al., 1997; Chen et al., 2009)。其中, 兜兰属是杓兰亚科最大的属, 约100多种, 占杓兰亚科总物种数一半以上(Govaerts et al., 2021)。兜兰属植物的基因组普遍较大并存在一定程度的变异(16.5-35.9 pg/C), 且染色体数目变异丰富(2n=26-42) (Leitch et al., 2009)。因此, 我们推测兜兰属可能存在全基因组复制事件, 然而, 在过去样本量小、跨大尺度的研究中并未检测到兜兰属特异发生的全基因组复制事件。因此, 本研究基于NCBI共享数据, 即杏黄兜兰(Paphiopedilum armeniacum)、同色兜兰(P. concolor)、带叶兜兰(P. hirsutissimum)以及麻栗坡兜兰(P. malipoense)的转录组数据, 采用经典的同义替换率(Ks)、系统发生基因组学以及相对定年的方法对其进行全基因组复制事件检测, 进而开展以下研究: (1) 过去未检测到全基因组复制事件历史的兜兰属植物是否发生了全基因组复制事件; (2) 若发生了全基因组复制事件, 进一步分析其发生时间, 以及是否为兜兰属内发生的全基因组复制事件; (3) 全基因组复制事件的发生对于兜兰属植物适应性演化的意义。
1 材料与方法
1.1 测序数据下载
从NCBI网站SRA数据库检索下载杏黄兜兰(Paphiopedilum armeniacum S.C.Chen & F.Y.Liu) (2n=26)、同色兜兰(P. concolor (Lindl. ex Bateman) Pfitzer) (2n=26)、带叶兜兰(P. hirsutissimum (Lindl. ex Hook.) Stein) (2n=26)以及麻栗坡兜兰(P. malipoense S.C. Chen & Z.H.Tsi) (2n=26)转录组测序的原始数据(raw data) (Cox et al., 1998; 杨志娟, 2006; Li et al., 2014; Zhang et al., 2017; Fang et al., 2020), 用于后续的组装与分析。同时, 从NCBI网站Genome数据库下载深圳拟兰(Apostasia shenzhenica Z.J.Liu & L.J. Chen)基因组数据(GCA_002786265.1) (Zhang et al., 2017)用于物种间直系同源基因的Ks分析。将拟兰作为基于系统发生基因组学检测全基因组复制事件的外类群。
1.2 测序数据提取和质控
借助SRA Toolkit v2.10.8中的fastq-dump命令从原始数据中提取获得fastq文件, 参数为--gzip --split-e (
1.3 转录组组装和质量评估
采用Trinity v2.11.0对高质量数据进行de novo组装(Haas et al., 2013), 参数设置为--seqType fq --min_ kmer_cov 2 --normalize_reads --bflyCalculateCPU。随后, 利用cd-hit v4.8.1将相似性≥95%的转录本(transcript)聚为一组(参数: -c 0.95), 每一组聚类中输出最长序列, 得到非冗余的单基因簇(unigene) (Li and Godzik, 2006)。基于embryophyta_odb10数据库, 利用BUSCO v4.0.6软件对组装获得的转录本进行完整性评估(Simão et al., 2015)。
1.4 蛋白编码区及转录因子预测
在默认设置条件下, 利用TransDecoder v5.5.0对unigene序列进行蛋白编码区预测(
1.5 全基因组复制事件检测
根据文献报道的方法计算物种内旁系同源基因对的Ks值(Sollars et al., 2017), 并对其进行正态分布拟合, 以检测全基因组复制事件。首先, 分别对各物种的蛋白序列进行all against all序列相似性比对(BLASTP), 阈值设置为e-5。然后, 应用脚本KSPlotter.py计算每个物种的Ks值(
为分析全基因组复制事件与类群分化间的时间关系, 利用wgd软件, 采用wf2流程分别计算4种兜兰与深圳拟兰、3种兜兰与杏黄兜兰(位于兜兰基部类群)间直系同源基因的Ks值(Zwaenepoel and Van De Peer, 2019)。若物种内旁系同源基因的Ks峰值(代表全基因组复制事件)小于物种间直系同源基因的Ks峰值(代表类群分化事件), 则认为全基因组复制事件发生在类群分化事件后; 反之, 则认为全基因组复制事件发生在类群分化事件前(One Thousand Plant Tran scriptomes Initiative, 2019)。
为验证Ks法检测结果的准确性, 应用tree2gd软件, 基于系统发生基因组学的方法再次检测全基因组复制事件(Zhang et al., 2020; Zhao et al., 2021)。(1) 以4种兜兰和深圳拟兰的蛋白序列为输入文件, 利用OrthoFinder v2.5.2筛选单拷贝直系同源基因(Emms and Kelly, 2019)。(2) 利用单拷贝直系同源基因构建物种树。首先, 采用MUSCLE v3.8.31对筛选得到的302个单拷贝直系同源基因进行多序列比对(Edgar, 2004); 随后, 基于比对结果使用Gblocks v0.91b筛选保守区域(Castresana, 2000; Talavera and Castresana, 2007), 并将筛选获得的保守区域串联形成多基因矩阵; 最后, 以ProtTest v3.4.2确定的PROTGAMMAJTTF为最优替代模型(Darriba et al., 2011), 利用RAxML v8.2.12软件, 采用最大似然法、基于保守序列矩阵、以深圳拟兰为外类群、在自举检验1 000次的设置下构建系统发生树(Stamatakis, 2014)。(3) 以第(2)步构建的系统发生树为物种树, 利用tree2gd v1.0.39软件, 基于默认参数检测全基因组复制事件(
1.6 全基因组复制事件相对定年
1.7 转录组功能注释和复制基因功能富集分析
首先, 分别对每种兜兰的所有蛋白序列进行功能注释。使用eggNOG-mapper v2.0.6软件, 基于eggNOG v5.0.1数据库对预测获得的蛋白序列进行功能注释(Huerta-Cepas et al., 2017, 2019), 注释结果用于分析保留复制基因的功能富集。然后, 根据高斯混合模型拟合显著存在的峰值, 分别提取4种兜兰各峰值95%置信区间的基因作为全基因组复制事件中保留的复制基因, 对其进行GO功能富集分析。借助R包AnnotationForge, 基于4种兜兰的功能注释结果, 为每个物种分别构建数据库(
2 结果与讨论
2.1 原始数据下载、组装和质量评估
从NCBI网站下载麻栗坡兜兰、同色兜兰、带叶兜兰以及杏黄兜兰的转录组原始数据, 用于测序的组织分别为茎、叶或种子, 原始数据量为3-14.1 Gb, 总数据量为28.7 Gb。对获得的原始数据进行提取、质控及组装, 分别组装得到76 006 (带叶兜兰) -239 105 (麻栗坡兜兰)个转录本, 去冗余后对应获得62 565-201 606个unigenes。具体信息见表1。
表1 转录组原始数据和de novo组装结果
Table 1
| Paphiopedilum concolor | P. hirsutissimum | P. malipoense | P. armeniacum | |
|---|---|---|---|---|
| Accession number | SRR1405683 | SRR1405685 | SRR5722160 | SRR9842184 |
| Tissues | Leaf | Leaf | Stem | Seed |
| Bases (Gb) | 3.6 | 3 | 14.1 | 8 |
| Number of transcripts | 156581 | 76006 | 239105 | 164515 |
| Average length of transcript (bp) | 907.1 | 1162.3 | 884.5 | 993.2 |
| N50 of transcript (bp) | 1486 | 1971 | 1627 | 1856 |
| Number of unigenes | 116919 | 62565 | 201606 | 139203 |
| Average length of unigene (bp) | 863.3 | 1071.1 | 815.6 | 906.4 |
| N50 of unigene (bp) | 1438 | 1829 | 1480 | 1704 |
| Source of raw data | Li et al., 2014 | Li et al., 2014 | Zhang et al., 2017 | Fang et al., 2020 |
为评估组装的完整性, 基于包含1 614个单拷贝基因的embryophyta_odb10数据库进行BUSCO评估, 完整覆盖基因的比例(complete BUSCOs)分别为麻栗坡兜兰(94.0%)>杏黄兜兰(92.5%)>同色兜兰(88.5%)>带叶兜兰(86.9%) (图1)。BUSCO评估结果显示, 组装完整性较高, 可用于后续分析。
图1
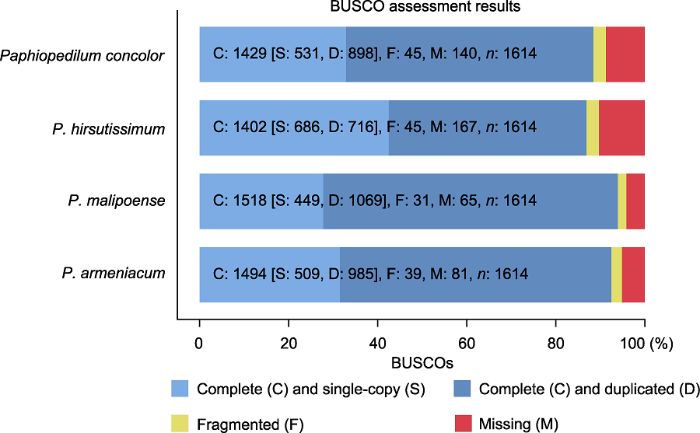
图1
BUSCO评估结果
C: 完整覆盖的基因数; S: 完整覆盖且为单拷贝的基因数; D: 完整覆盖且为多拷贝的基因数; F: 未完整覆盖的基因数; M: 缺失基因数
Figure 1
BUSCO assessment results
C: Complete BUSCOs; S: Complete and single-copy BUSCOs; D: Complete and duplicated BUSCOs; F: Fragmented BUSCOs; M: Missing BUSCOs
2.2 蛋白编码区和转录因子预测
表2 蛋白编码区和转录因子预测结果
Table 2
| Paphiopedilum concolor | P. hirsutissimum | P. malipoense | P. armeniacum | |
|---|---|---|---|---|
| Number of protein coding sequences | 56439 | 33207 | 79854 | 58575 |
| Average length of protein coding sequence (bp) | 936.1 | 994.9 | 829.1 | 914.7 |
| N50 of protein coding sequence (bp) | 1209 | 1308 | 1089 | 1215 |
| Number of CDS identified as transcription factor | 1950 | 1181 | 2586 | 2014 |
| Number of transcription factor families | 66 | 67 | 67 | 68 |
2.3 全基因组复制事件检测
图2
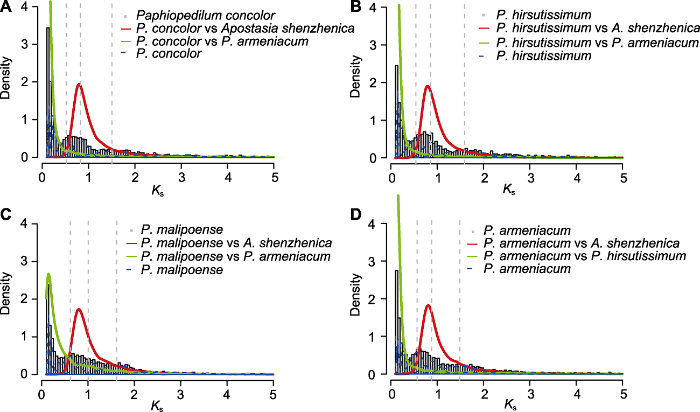
图2
4种兜兰的Ks密度分布
灰色直方图: 物种内旁系同源基因的Ks密度分布; 红色曲线: 4种兜兰与深圳拟兰间直系同源基因的Ks密度分布; 绿色曲线: 杏黄兜兰与其它3种兜兰间直系同源基因的Ks密度分布; 蓝色虚线: 物种内旁系同源基因Ks值的高斯混合模型拟合结果; 灰色虚线: 高斯混合模型显著拟合到的Ks峰值。
Figure 2
The density plot of Ks from four species of Paphiopedilum
Histograms filled in grey: The density distributions of intraspecies paralogue Ks values; Red solid curves: The Ks density plots of interspecies orthologues between four species of Paphiopedilum and Apostasia shenzhenica; Green solid curves: The Ks density plots of interspecies orthologues between P. armeniacum and other three species of Paphiopedilum; Blue dashed curves: The fitting results based on Gaussian mixture modeling of intraspecies paralogue Ks values; Grey dashed lines: The Ks values of significant peaks identified by Gaussian mixture modeling.
表3 基于高斯混合模型拟合的Ks结果
Table 3
| Species | No. of components | No. of duplicates | BIC | Variance | Mean (Ks) | Proportion |
|---|---|---|---|---|---|---|
| Paphiopedilum concolor | 9 | 207 | -4673.731 | 0.0000 | 0.1077 | 0.0527 |
| 9 | 385 | -4673.731 | 0.0002 | 0.1314 | 0.1048 | |
| 9 | 416 | -4673.731 | 0.0007 | 0.1740 | 0.1194 | |
| 9 | 311 | -4673.731 | 0.0026 | 0.2517 | 0.0964 | |
| 9 | 522 | -4673.731 | 0.0175 | 0.5161 | 0.1544 | |
| 9 | 642 | -4673.731 | 0.0241 | 0.8236 | 0.1741 | |
| 9 | 567 | -4673.731 | 0.1557 | 1.5043 | 0.1594 | |
| 9 | 300 | -4673.731 | 0.5765 | 2.4529 | 0.1148 | |
| 9 | 90 | -4673.731 | 0.1277 | 4.3036 | 0.0240 | |
| P. hirsutissimum | 7 | 196 | -4604.688 | 0.0001 | 0.1146 | 0.0632 |
| 7 | 277 | -4604.688 | 0.0005 | 0.1504 | 0.0991 | |
| 7 | 290 | -4604.688 | 0.0026 | 0.2292 | 0.1109 | |
| 7 | 558 | -4604.688 | 0.0282 | 0.5407 | 0.2162 | |
| 7 | 508 | -4604.688 | 0.0260 | 0.8544 | 0.1693 | |
| 7 | 585 | -4604.688 | 0.2594 | 1.5894 | 0.2351 | |
| 7 | 236 | -4604.688 | 0.8550 | 3.0527 | 0.1062 | |
| Species | No. of components | No. of duplicates | BIC | Variance | Mean (Ks) | Proportion |
| P. malipoense | 9 | 377 | -14027.68 | 0.0000 | 0.1081 | 0.0399 |
| 9 | 751 | -14027.68 | 0.0003 | 0.1362 | 0.0890 | |
| 9 | 820 | -14027.68 | 0.0016 | 0.2006 | 0.1005 | |
| 9 | 579 | -14027.68 | 0.0067 | 0.3290 | 0.0768 | |
| 9 | 1611 | -14027.68 | 0.0287 | 0.6196 | 0.1983 | |
| 9 | 1464 | -14027.68 | 0.0447 | 1.0026 | 0.1778 | |
| 9 | 1634 | -14027.68 | 0.1002 | 1.6186 | 0.1952 | |
| 9 | 683 | -14027.68 | 0.5901 | 2.6205 | 0.1105 | |
| 9 | 105 | -14027.68 | 0.0568 | 4.5773 | 0.0121 | |
| P. armeniacum | 9 | 206 | -8261.607 | 0.0000 | 0.1063 | 0.0359 |
| 9 | 472 | -8261.607 | 0.0002 | 0.1296 | 0.0870 | |
| 9 | 478 | -8261.607 | 0.0008 | 0.1744 | 0.0950 | |
| 9 | 400 | -8261.607 | 0.0039 | 0.2729 | 0.0849 | |
| 9 | 1089 | -8261.607 | 0.0213 | 0.5664 | 0.2105 | |
| 9 | 778 | -8261.607 | 0.0287 | 0.8825 | 0.1457 | |
| 9 | 999 | -8261.607 | 0.1443 | 1.4888 | 0.1980 | |
| 9 | 455 | -8261.607 | 0.5375 | 2.4393 | 0.1195 | |
| 9 | 120 | -8261.607 | 0.1481 | 4.3256 | 0.0234 |
BIC: 贝叶斯信息标准
BIC: Bayesian information criterion
除计算物种内旁系同源基因的Ks值外, 我们还分别计算了4种兜兰与兰科基部代表物种深圳拟兰、3种兜兰与兜兰基部代表物种杏黄兜兰间直系同源基因的Ks值(图2), 结果发现4种兜兰与深圳拟兰间直系同源基因的Ks峰值均小于全基因组复制事件WGD1和WGD2的Ks峰值, 大于全基因组复制事件WGD3的Ks峰值, 推测全基因组复制事件WGD1和WGD2发生在兜兰属与深圳拟兰分化事件之前, 而WGD3则发生在之后。杏黄兜兰与其它3种兜兰间直系同源基因的Ks峰值均小于全基因组复制事件WGD1、WGD2和WGD3, 推测3次全基因组复制事件均发生在兜兰属分化之前。
应用tree2gd v1.0.39软件, 基于系统发生基因组学方法再次进行全基因组复制事件检测, 判定标准参照Zhang等(2020)所述方法。满足以下任一条件则认为发生了全基因组复制事件: (1) 复制基因(gene duplication, GD) >500个, 其中(AB)(AB)类型的复制基因>250个; (2) 复制基因>1 500个, 其中(AB)(AB)类型的复制基因>100个, 且(AB)(AB)类型的复制基因与(AB)A类型或(AB)B类型的复制基因之和>1 000个。tree2gd分析结果(图3)表明, 4种兜兰的祖先(即结点2)保留了556个复制基因, 其中(AB)(AB)类型的复制基因为274个, 满足全基因组复制事件的判定条件, 推测在兜兰属与深圳拟兰分化之后、兜兰属分化之前(即结点3与结点2之间)发生了1次全基因组复制事件,与采用Ks检测到的WGD3相一致。由于tree2gd软件主要基于系统发生基因组学方法进行检测, 因此受样本限制(仅4种兜兰和深圳拟兰)无法检测到兜兰属以外的全基因组复制事件。
图3
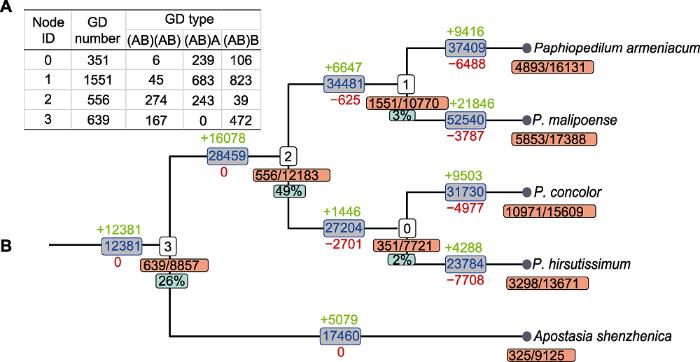
图3
基于系统发生基因组学的全基因组复制检测结果
(A) 各结点的复制基因家族情况统计, 其中Node ID对应(B)中相应结点; GD number为各结点复制基因家族数量; GD type为复制基因家族每种类型的数量; (B) 结点下方黄色方框内数字为复制基因家族的数量/基因家族的数量, 绿色方框内数字为(AB)(AB)类型复制基因家族占复制基因家族的比例; 分支上方绿色数字、下方红色数字分别表示基因家族的扩张和收缩。
Figure 3
The detection of whole-genome duplication based on phylogenomics
(A) The statistics of duplicated gene families, Node ID corresponds to the node number in (B); GD number is the number of duplicated gene families at each node; GD type is the number of each type of duplicated gene families; (B) The numbers in yellow box below nodes is the number of duplicated gene families/gene families, the corresponding green box is the percentage of (AB)(AB) types; numbers above (green) and below (red) branches indicate the expansion and contraction of gene families, respectively.
2.4 全基因组复制事件的相对定年
根据高斯混合模型拟合到的Ks峰, 采用深圳拟兰r=6.76×10-9 (同义替换/位点/年), 计算4种兜兰发生3次全基因组复制事件的时间, 结果分别为110.17-119.77、60.95-74.19和38.19-45.85 Mya (表4)。
表4 基于Ks峰值对全基因组复制(WGD)事件进行定年
Table 4
| Species | Name of WGD | Mean (Ks) | Age of WGD calculated by Ks mean value (Mya) | Age of WGD with 95% confidence interval (Mya) |
|---|---|---|---|---|
| Paphiopedilum concolor | WGD3 | 0.5161 | 38.19 | 37.35-39.03 |
| WGD2 | 0.8236 | 60.95 | 60.06-61.83 | |
| WGD1 | 1.5043 | 111.32 | 108.91-113.72 | |
| P. hirsutissimum | WGD3 | 0.5407 | 40.01 | 38.98-41.04 |
| WGD2 | 0.8544 | 63.22 | 62.19-64.26 | |
| WGD1 | 1.5894 | 117.61 | 114.56-120.67 | |
| P. malipoense | WGD3 | 0.6196 | 45.85 | 45.24-46.46 |
| WGD2 | 1.0026 | 74.19 | 73.39-74.99 | |
| WGD1 | 1.6186 | 119.77 | 118.64-120.91 | |
| P. armeniacum | WGD3 | 0.5664 | 41.92 | 41.28-42.56 |
| WGD2 | 0.8825 | 65.31 | 64.43-66.19 | |
| WGD1 | 1.4888 | 110.17 | 108.42-111.91 |
2.5 转录组功能注释和复制基因的功能富集分析
对4种兜兰的CDS序列进行功能注释, 结果显示, 共有102 214个CDS注释到GO数据库(包括同色兜兰27 528个, 带叶兜兰15 721个, 麻栗坡兜兰33 522个, 杏黄兜兰25 443个), 占CDS总数的44.82%。对4种兜兰的功能注释结果进行二级分类统计(图4), 结果共注释到26个生物学过程(biological process, BP)、4种细胞组分(cellular component, CC)及17类分子功能(molecular function, MF)。在生物学过程中, 参与细胞进程(cellular process)和代谢过程(metabolic process)的基因数量最多; 在细胞组分中, 表达细胞解剖实体(cellular anatomical entity)和含蛋白复合体组分(protein-containing complex)的基因数量最多; 在分子功能中, 参与催化活性(catalytic activity)和结合功能(binding)的基因数量最多。
图4

图4
转录组GO功能注释和复制基因功能富集的二级分类统计
Transcriptome: 转录组功能注释结果; WGD1: 全基因组复制事件WGD1中复制基因的功能富集结果(P<0.05); WGD2: 全基因组复制事件WGD2中复制基因的功能富集结果(P<0.05); WGD3: 全基因组复制事件WGD3中复制基因的功能富集结果(P<0.05); BP: 生物学过程; CC: 细胞组分; MF: 分子功能
Figure 4
The level 2 GO categories of transcriptome functional annotation and duplicated gene functional enrichment
Transcriptome: Results of transcriptome functional annotation; WGD1: Functional enrichment of duplicated gene from whole- genome duplication (WGD) event WGD1 (P<0.05); WGD2: Functional enrichment of duplicated gene from WGD2 (P<0.05); WGD3: Functional enrichment of duplicated gene from WGD3 (P<0.05); BP: Biological process; CC: Cellular component; MF: Molecular function
分别提取4种兜兰各全基因组复制事件保留的复制基因进行GO功能富集分析, 发现有1 694个GO条目得到显著富集(P<0.05)。基于GO条目间的关系, 在二级节点对其进行分类统计(图4), 共富集到20个生物学过程、2种细胞组分和12类分子功能; 其中, 70%生物学过程、100%细胞组分和50%分子功能在3次全基因组复制事件中均得到富集。对3次全基因组复制事件进行比较分析, 发现WGD1无特异富集的生物学过程; WGD2特异富集了节律过程(rhythmic process)和移动(locomotion)的生物学过程; WGD3特异富集了色素沉积(pigmentation)、种内个体间相互作用(intraspecies interaction between organisms)和反应(behavior)的生物学过程。
为分析各全基因组复制事件的功能保留模式, 我们提取4种兜兰中3种及以上物种共有的功能进行比较分析(图5), 发现在WGD1共同富集了36个GO条目, 包括脂类代谢、单糖代谢、苯丙烷类的合成与代谢、叶片发育和衰老调控、活性氧代谢过程调控等生物学过程, 以及氧化还原酶活性等分子功能。相较其它2次全基因组复制事件, WGD1特异富集了次生代谢过程、单糖代谢过程、叶片发育和衰老调控以及受伤的应激反应等生物学过程。在WGD2共同富集了3个GO条目, 仅包括脱落酸激活的信号通路1个生物学过程。在WGD3共同富集了40个GO条目, 包括酶联受体蛋白信号通路、根表皮细胞分化, 以及特异富集了行为、植物表皮和保卫细胞的形态发生、毛状体分化、细胞壁β-葡聚糖和纤维素代谢过程、甘油酯和甘油磷脂代谢过程、细胞防御反应、色素沉着和细胞发育的正向调控等生物学过程。
图5
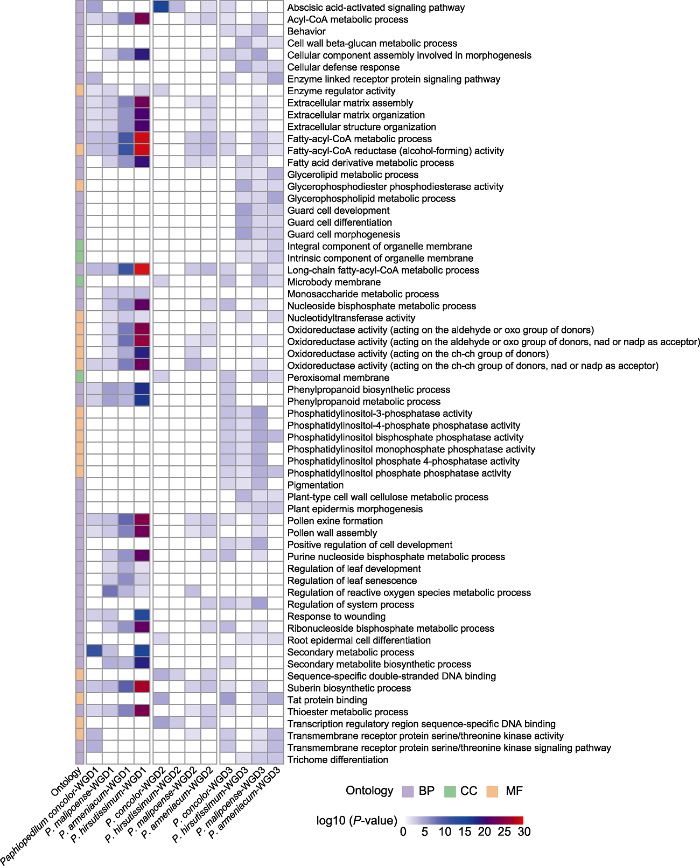
图5
基于GO功能注释结果的3种及以上兜兰物种共同富集的GO条目
BP、CC和MF同
Figure 5
The shared GO terms of 3 or more Paphiopedilum species based on the results of GO functional enrichment
BP, CC, and MF see
2.6 讨论
2.6.1 兰科植物的全基因组复制历史
目前, 在兰科植物中仅检测到2次全基因组复制事件, 一次为大多数单子叶植物共享(110-135 Mya), 另一次为现存兰科植物共享(72-78 Mya)(Cai et al., 2015; Ming et al., 2015; Zhang et al., 2016, 2017; Yuan et al., 2018; One Thousand Plant Transcriptomes Initiative, 2019; Hasing et al., 2020)。兜兰属是兰科多样性的重要代表类群, 本研究基于4种兜兰的转录组数据, 检测到3次全基因组复制事件, 分别发生在110.17-119.77 Mya (WGD1)、60.95-74.19 Mya (WGD2)和38.19-45.85 Mya (WGD3)。其中, WGD1和WGD2发生时间与前期研究得出的2次全基因组复制事件相近, 且物种间Ks分析表明, 二者均发生在兜兰属与深圳拟兰分化事件之前(图2), 因此推测WGD1为大多数单子叶植物共享、WGD2为现存兰科植物共享的全基因组复制事件。而本研究中检测到的全基因组复制事件WGD3 (38.19-45.85 Mya), 在蓝莓(blueberry)、茶树(Camellia sinensis var. sinensis)和胡萝卜(Daucus carota)中同一时期也检测到了全基因组复制事件(Iorizzo et al., 2016; Wei et al., 2018; Wang et al., 2020), 豆科中更是在该段时间检测到大量全基因组复制事件(17次, 23-55 Mya) (Zhao et al., 2021), 但在兰科植物中尚未见报道。
综合类群分化的时间信息、物种间Ks检测结果以及tree2gd检测结果, 进一步分析WGD3在兰科中的系统发生位置。兰科5个亚科的亲缘关系为(拟兰亚科(香荚兰亚科(杓兰亚科(兰亚科, 树兰亚科)))), 其中杓兰亚科与姐妹类群的分化时间约为64.97 Mya (48.54-84.93 Mya) (Kim et al., 2020), 冠群时间为33 Mya (19-50 Mya) (Gustafsson et al., 2010), 而WGD3的发生时间为38.19-45.85 Mya, 初步推测WGD3为杓兰亚科特异发生的全基因组复制事件。杓兰亚科包含5个属, 其亲缘关系为(杓兰属(南美杓兰属(兜兰属(美洲兜兰属, 镊萼兜兰属)))), 兜兰属与姐妹类群的分化时间为29.9 Mya (14.6-39.1 Mya) (
2.6.2 全基因组复制事件对兜兰属植物适应性演化的意义
多倍化或全基因组复制, 特别是在稳定环境下, 常被认为是进化的终点(Comai, 2005; Oberlander et al., 2016)。然而, 在植物的演化过程中, 全基因组复制并非随机发生, 而是与全球气候变化、地质变化或者大规模灭绝等密切相关, 发生全基因组复制的个体在胁迫或极端环境条件下具有较二倍体祖先更强的适应性(Van de Peer et al., 2017, 2021; Ren et al., 2018; Wu et al., 2020)。与上述研究结果相似, 本研究检测到的3次全基因组复制事件发生时期出现了全球气候变化或大规模灭绝事件, 推测全基因组复制事件提高了兜兰属植物祖先应对极端环境变化的适应性。例如, WGD1 (110.17-119.77 Mya)发生在白垩纪(Cretaceous)阿普特阶(Aptian)至阿尔布阶(Albian), 随后出现了超级温室期(83.6-93.9 Mya) (Klages et al., 2020); WGD2 (60.95-74.19 Mya)发生在白垩纪与古近纪(Paleogene)交界, 出现了白垩纪-古近纪灭绝事件(K-Pg灭绝事件) (Vellekoop et al., 2016); WGD3 (38.19-45.85 Mya)发生在古近纪始新世(Eocene), 发生了古新世-始新世极热事件(56 Mya)和始新世-渐新世(Oligocene)全球变冷(Zachos et al., 2001; McInerney and Wing, 2011)。
全基因组复制事件保留了部分复制基因, 对保留的复制基因进行功能分析可为阐明全基因组复制事件对植物适应性演化的促进作用提供遗传证据。本研究分别对4种兜兰3次全基因组复制后的保留复制基因进行了GO功能富集分析, 发现3次全基因组复制事件富集到的功能存在差异(图4, 图5)。WGD1富集了脂类代谢、软木脂的生物合成、苯丙烷类的合成与代谢, 以及氧化还原酶活性和活性氧代谢过程的调控等功能(图5), 这可能与兜兰属植物应对超级温室期的干旱环境以及抵御干旱引起的活性氧失衡有关(Upchurch, 2008; Das and Roychoudhury, 2014; Brunner et al., 2015; Zhang et al., 2021b)。在K-Pg灭绝时期, 大气中充满了灰尘、硫酸盐气溶胶及碳黑颗粒, 黑暗和低温成为主要的胁迫因子(Vellekoop et al., 2016)。推测WGD2富集的脱落酸激活的信号通路以及昼夜节律等功能提高了兜兰属植物祖先对当时剧变环境的适应性(图4, 图5) (杨有新等, 2014; Vishwakarma et al., 2017)。WGD3之后, 兜兰属植物祖先经历了全球温度骤降, 推测富集的磷脂代谢、酶联受体蛋白信号通路、色素沉着, 以及保卫细胞分化与发育、根表皮细胞分化与毛状体分化等功能, 可能与应对低温引起的植物萎蔫、叶绿素含量减少以及细胞膜发生相变有关(王芳等, 2019)。综上, 推测保留的复制基因在功能上与当时特定的胁迫因子相关。
上述分析结果与前人有关被子植物主要分支的研究结论一致(Wu et al., 2020)。Wu等(2020)对包括被子植物主要分支在内的25个物种(双子叶植物10种, 单子叶植物12种, 基部被子植物、石松类植物和苔藓各1种)在3个历史时期(约120 Mya、约66 Mya及<20 Mya)发生全基因组复制后的保留复制基因进行了功能富集分析, 发现不同时期多倍化后的保留复制基因在功能上与当时的环境压力一致。(1) 在约120 Mya发生全基因组复制事件后的复制基因主要在响应缺水和盐胁迫的功能上显著富集, 当时地球正处于干旱环境; (2) 在K-Pg灭绝时期(约66 Mya)发生了全球变冷、黑暗、酸雨和野火等, 该时期富集到了与冷、热、渗透、盐和水等胁迫相关的功能, 以及脱落酸激活的信号通路等与胁迫响应相关的其它生物学过程; (3) 在约20 Mya发生全基因组复制事件后的复制基因主要在响应盐胁迫、缺水和机械伤害的功能上显著富集, 与当时CO2浓度低和相对低温有关。
附表1 4种兜兰转录因子家族
Appendix table 1 Transcription factor family in the four Paphiopedilum species
参考文献

全基因组复制在动植物中普遍存在, 被认为是促进物种进化的重要动力之一。作为蕨类植物的单种科物种, 翼盖蕨(Didymochlaena trancatula)是真水龙骨类I的基部类群, 在蕨类中具有独特的演化地位。本研究基于高通量测序, 通过同义替换率(Ks)分析、相对定年分析揭示翼盖蕨的全基因组复制发生情况。Ks分析表明, 翼盖蕨至少经历了两次全基因组复制事件, 其中一次发生于59-62 million years ago (Mya), 另一次发生于90-94 Mya, 这两次全基因组复制事件分别和白垩纪第三纪的Cretaceous-Tertiary (C-T)大灭绝事件以及翼盖蕨的物种分化时间相吻合。进一步对两次全基因组复制保留的基因进行功能注释和富集分析, 结果显示与转录及代谢调控相关的基因优势被保留。翼盖蕨的全基因组复制事件可能促进了该物种的分化及其对极端环境的适应性。
The ongoing climate change is characterized by increased temperatures and altered precipitation patterns. In addition, there has been an increase in both the frequency and intensity of extreme climatic events such as drought. Episodes of drought induce a series of interconnected effects, all of which have the potential to alter the carbon balance of forest ecosystems profoundly at different scales of plant organization and ecosystem functioning. During recent years, considerable progress has been made in the understanding of how aboveground parts of trees respond to drought and how these responses affect carbon assimilation. In contrast, processes of belowground parts are relatively underrepresented in research on climate change. In this review, we describe current knowledge about responses of tree roots to drought. Tree roots are capable of responding to drought through a variety of strategies that enable them to avoid and tolerate stress. Responses include root biomass adjustments, anatomical alterations, and physiological acclimations. The molecular mechanisms underlying these responses are characterized to some extent, and involve stress signaling and the induction of numerous genes, leading to the activation of tolerance pathways. In addition, mycorrhizas seem to play important protective roles. The current knowledge compiled in this review supports the view that tree roots are well equipped to withstand drought situations and maintain morphological and physiological functions as long as possible. Further, the reviewed literature demonstrates the important role of tree roots in the functioning of forest ecosystems and highlights the need for more research in this emerging field.
The use of some multiple-sequence alignments in phylogenetic analysis, particularly those that are not very well conserved, requires the elimination of poorly aligned positions and divergent regions, since they may not be homologous or may have been saturated by multiple substitutions. A computerized method that eliminates such positions and at the same time tries to minimize the loss of informative sites is presented here. The method is based on the selection of blocks of positions that fulfill a simple set of requirements with respect to the number of contiguous conserved positions, lack of gaps, and high conservation of flanking positions, making the final alignment more suitable for phylogenetic analysis. To illustrate the efficiency of this method, alignments of 10 mitochondrial proteins from several completely sequenced mitochondrial genomes belonging to diverse eukaryotes were used as examples. The percentages of removed positions were higher in the most divergent alignments. After removing divergent segments, the amino acid composition of the different sequences was more uniform, and pairwise distances became much smaller. Phylogenetic trees show that topologies can be different after removing conserved blocks, particularly when there are several poorly resolved nodes. Strong support was found for the grouping of animals and fungi but not for the position of more basal eukaryotes. The use of a computerized method such as the one presented here reduces to a certain extent the necessity of manually editing multiple alignments, makes the automation of phylogenetic analysis of large data sets feasible, and facilitates the reproduction of the final alignment by other researchers.
Polyploids - organisms that have multiple sets of chromosomes - are common in certain plant and animal taxa, and can be surprisingly stable. The evidence that has emerged from genome analyses also indicates that many other eukaryotic genomes have a polyploid ancestry, suggesting that both humans and most other eukaryotes have either benefited from or endured polyploidy. Studies of polyploids soon after their formation have revealed genetic and epigenetic interactions between redundant genes. These interactions can be related to the phenotypes and evolutionary fates of polyploids. Here, I consider the advantages and challenges of polyploidy, and its evolutionary potential.
Nuclear DNA contents (4C) were estimated by Feulgen microdensitometry in 27 species of slipper orchids. These data and recent information concerning the molecular systematics of Cypripedioideae allow an interesting re-evaluation of karyotype and genome size variation among slipper orchids in a phylogenetic context. DNA amounts differed 5.7-fold, from 24.4 pg in Phragmipedium longifolium to 138.1 pg in Paphiopedilum wardii. The most derived clades of the conduplicate-leaved slipper orchids have undergone a radical process of genome fragmentation that is most parsimoniously explained by Robertsonian changes involving centric fission. This process seems to have occurred independently of genome size variation. However, it may reflect environmental or selective pressures favoring higher numbers of linkage groups in the karyotype.
We have implemented a high-performance computing (HPC) version of ProtTest that can be executed in parallel in multicore desktops and clusters. This version, called ProtTest 3, includes new features and extended capabilities.ProtTest 3 source code and binaries are freely available under GNU license for download from http://darwin.uvigo.es/software/prottest3, linked to a Mercurial repository at Bitbucket (https://bitbucket.org/).dposada@uvigo.esSupplementary data are available at Bioinformatics online.
Detection of protein families in large databases is one of the principal research objectives in structural and functional genomics. Protein family classification can significantly contribute to the delineation of functional diversity of homologous proteins, the prediction of function based on domain architecture or the presence of sequence motifs as well as comparative genomics, providing valuable evolutionary insights. We present a novel approach called TRIBE-MCL for rapid and accurate clustering of protein sequences into families. The method relies on the Markov cluster (MCL) algorithm for the assignment of proteins into families based on precomputed sequence similarity information. This novel approach does not suffer from the problems that normally hinder other protein sequence clustering algorithms, such as the presence of multi-domain proteins, promiscuous domains and fragmented proteins. The method has been rigorously tested and validated on a number of very large databases, including SwissProt, InterPro, SCOP and the draft human genome. Our results indicate that the method is ideally suited to the rapid and accurate detection of protein families on a large scale. The method has been used to detect and categorise protein families within the draft human genome and the resulting families have been used to annotate a large proportion of human proteins.
Paphiopedilum is an important genus of the orchid family Orchidaceae and has high horticultural value. The wild populations are under threat of extinction because of overcollection and habitat destruction. Mature seeds of most Paphiopedilum species are difficult to germinate, which severely restricts their germplasm conservation and commercial production. The factors inhibiting germination are largely unknown.In this study, large amounts of non-methylated lignin accumulated during seed maturation of Paphiopedilum armeniacum (P. armeniacum), which negatively correlates with the germination rate. The transcriptome profiles of P. armeniacum seed at different development stages were compared to explore the molecular clues for non-methylated lignin synthesis. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed that a large number of genes associated with phenylpropanoid biosynthesis and phenylalanine metabolism during seed maturation were differentially expressed. Several key genes in the lignin biosynthetic pathway displayed different expression patterns during the lignification process. PAL, 4CL, HCT, and CSE upregulation was associated with C and H lignin accumulation. The expression of CCoAOMT, F5H, and COMT were maintained at a low level or down-regulated to inhibit the conversion to the typical G and S lignin. Quantitative real-time RT-PCR analysis confirmed the altered expression levels of these genes in seeds and vegetative tissues.This work demonstrated the plasticity of natural lignin polymer assembly in seed and provided a better understanding of the molecular mechanism of seed-specific lignification process.
A codon-based model for the evolution of protein-coding DNA sequences is presented for use in phylogenetic estimation. A Markov process is used to describe substitutions between codons. Transition/transversion rate bias and codon usage bias are allowed in the model, and selective restraints at the protein level are accommodated using physicochemical distances between the amino acids coded for by the codons. Analyses of two data sets suggest that the new codon-based model can provide a better fit to data than can nucleotide-based models and can produce more reliable estimates of certain biologically important measures such as the transition/transversion rate ratio and the synonymous/nonsynonymous substitution rate ratio.
Background: The temporal origin and diversification of orchids (family Orchidaceae) has been subject to intense debate in the last decade. The description of the first reliable fossil in 2007 enabled a direct calibration of the orchid phylogeny, but little attention has been paid to the potential influence of dating methodology in obtaining reliable age estimates. Moreover, two new orchid fossils described in 2009 have not yet been incorporated in a molecular dating analysis. Here we compare the ages of major orchid clades estimated under two widely used methods, a Bayesian relaxed clock implemented in BEAST and Penalized Likelihood implemented in r8s. We then perform a new family-level analysis by integrating all 3 available fossils and using BEAST. To evaluate how the newly estimated ages may influence the evolutionary interpretation of a species-level phylogeny, we assess divergence times for the South American genus Hoffmannseggella (subfam. Epidendroideae), for which we present an almost complete phylogeny (40 out of 41 species sampled). Results: Our results provide additional support that all extant orchids shared a most recent common ancestor in the Late Cretaceous (similar to 77 million years ago, Ma). However, we estimate the crown age of the five orchid subfamilies to be generally (similar to 1-8 Ma) younger than previously calculated under the Penalized Likelihood algorithm and using a single internal fossil calibration. The crown age of Hoffmannseggella is estimated here at similar to 11 Ma, some 3 Ma more recently than estimated under Penalized Likelihood. Conclusions: Contrary to recent suggestions that orchid diversification began in a period of global warming, our results place the onset of diversification of the largest orchid subfamilies (Orchidoideae and Epidendroideae) in a period of global cooling subsequent to the Early Eocene Climatic Optimum. The diversification of Hoffmannseggella appears even more correlated to late Tertiary climatic fluctuations than previously suggested. With the incorporation of new fossils in the orchid phylogeny and the use of a method that is arguably more adequate given the present data, our results represent the most up-to-date estimate of divergence times in orchids.
Orthology assignment is ideally suited for functional inference. However, because predicting orthology is computationally intensive at large scale, and most pipelines are relatively inaccessible (e.g., new assignments only available through database updates), less precise homology-based functional transfer is still the default for (meta-)genome annotation. We, therefore, developed eggNOG-mapper, a tool for functional annotation of large sets of sequences based on fast orthology assignments using precomputed clusters and phylogenies from the eggNOG database. To validate our method, we benchmarked Gene Ontology (GO) predictions against two widely used homology-based approaches: BLAST and InterProScan. Orthology filters applied to BLAST results reduced the rate of false positive assignments by 11%, and increased the ratio of experimentally validated terms recovered over all terms assigned per protein by 15%. Compared with InterProScan, eggNOG-mapper achieved similar proteome coverage and precision while predicting, on average, 41 more terms per protein and increasing the rate of experimentally validated terms recovered over total term assignments per protein by 35%. EggNOG-mapper predictions scored within the top-5 methods in the three GO categories using the CAFA2 NK-partial benchmark. Finally, we evaluated eggNOG-mapper for functional annotation of metagenomics data, yielding better performance than interProScan. eggNOG-mapper runs ∼15× faster than BLAST and at least 2.5× faster than InterProScan. The tool is available standalone and as an online service at http://eggnog-mapper.embl.de.© The Author 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Whole-genome duplications are widespread across land plant phylogenies and particularly frequent in ferns and angiosperms. Genome duplications spurred the evolution of key innovations associated with diversification in many angiosperm clades and lineages. Such diversifications are not initiated by genome doubling per se. Rather, differentiation of the primary polyploid populations through a range of processes results in post-polyploid genome diploidization. Structural diploidization gradually reverts the polyploid genome to one functionally diploid-like through chromosomal rearrangements which frequently result in dysploid changes. Dysploidies may lead to reproductive isolation among post-polyploid offspring and significantly contribute to speciation and cladogenetic events.Copyright © 2018 Elsevier Ltd. All rights reserved.
Pineapple (Ananas comosus (L.) Merr.) is the most economically valuable crop possessing crassulacean acid metabolism (CAM), a photosynthetic carbon assimilation pathway with high water-use efficiency, and the second most important tropical fruit. We sequenced the genomes of pineapple varieties F153 and MD2 and a wild pineapple relative, Ananas bracteatus accession CB5. The pineapple genome has one fewer ancient whole-genome duplication event than sequenced grass genomes and a conserved karyotype with seven chromosomes from before the ρ duplication event. The pineapple lineage has transitioned from C3 photosynthesis to CAM, with CAM-related genes exhibiting a diel expression pattern in photosynthetic tissues. CAM pathway genes were enriched with cis-regulatory elements associated with the regulation of circadian clock genes, providing the first cis-regulatory link between CAM and circadian clock regulation. Pineapple CAM photosynthesis evolved by the reconfiguration of pathways in C3 plants, through the regulatory neofunctionalization of preexisting genes and not through the acquisition of neofunctionalized genes via whole-genome or tandem gene duplication.
We describe here the reconstruction of the genome of the most recent common ancestor (MRCA) of modern monocots and eudicots, accounting for 95% of extant angiosperms, with its potential repertoire of 22,899 ancestral genes conserved in present-day crops. The MRCA provides a starting point for deciphering the reticulated evolutionary plasticity between species (rapidly versus slowly evolving lineages), subgenomes (pre-versus post-duplication blocks), genomic compartments (stable versus labile loci), genes (ancestral versus species-specific genes) and functions (gained versus lost ontologies), the key mutational forces driving the success of polyploidy in crops. The estimation of the timing of angiosperm evolution, based on MRCA genes, suggested that this group emerged 214 million years ago during the late Triassic era, before the oldest recorded fossil. Finally, the MRCA constitutes a unique resource for scientists to dissect major agronomic traits in translational genomics studies extending from model species to crops.
Whole-genome duplication (WGD) in angiosperms has been hypothesized to be advantageous in unstable environments and/or to increase diversification rates, leading to radiations. Under the first hypothesis, floras in stable environments are predicted to have lower proportions of polyploids than highly, recently disturbed floras, whereas species-rich floras would be expected to have higher than expected proportions of polyploids under the second. The South African Cape flora is used to discriminate between these two hypotheses because it features a hyperdiverse flora predominantly generated by a limited number of radiations (Cape clades), against a backdrop of climatic and geological stability.We compiled all known chromosome counts for species in 21 clades present in the Cape (1653 species, including 24 Cape clades), inferred ploidy levels for these species by inspection or derived from the primary literature, and compared Cape to non-Cape ploidy levels in these clades (17,520 species) using G tests.The Cape flora has anomalously low proportions of polyploids compared with global levels. This pattern is consistently observed across nearly half the clades and across global latitudinal gradients, although individual lineages seem to be following different paths to low levels of WGD and to differing degrees.This pattern shows that the diversity of the Cape flora is the outcome of primarily diploid radiations and supports the hypothesis that WGD may be rare in stable environments.© 2016 Botanical Society of America.
Gene duplications provide evolutionary potentials for generating novel functions, while polyploidization or whole genome duplication (WGD) doubles the chromosomes initially and results in hundreds to thousands of retained duplicates. WGDs are strongly supported by evidence commonly found in many species-rich lineages of eukaryotes, and thus are considered as a major driving force in species diversification. We performed comparative genomic and phylogenomic analyses of 59 public genomes/transcriptomes and 46 newly sequenced transcriptomes covering major lineages of angiosperms to detect large-scale gene duplication events by surveying tens of thousands of gene family trees. These analyses confirmed most of the previously reported WGDs and provided strong evidence for novel ones in many lineages. The detected WGDs supported a model of exponential gene loss during evolution with an estimated half-life of approximately 21.6 million years, and were correlated with both the emergence of lineages with high degrees of diversification and periods of global climate changes. The new datasets and analyses detected many novel WGDs widely spread during angiosperm evolution, uncovered preferential retention of gene functions in essential cellular metabolisms, and provided clues for the roles of WGD in promoting angiosperm radiation and enhancing their adaptation to environmental changes.Copyright © 2018 The Author. Published by Elsevier Inc. All rights reserved.
Using plant EST collections, we obtained 1392 potential gene duplicates across 8 plant species: Zea mays, Oryza sativa, Sorghum bicolor, Hordeum vulgare, Solanum tuberosum, Lycopersicon esculentum, Medicago truncatula, and Glycine max. We estimated the synonymous and nonsynonymous distances between each gene pair and identified two to three mixtures of normal distributions corresponding to one to three rounds of genome duplication in each species. Within the Poaceae, we found a conserved duplication event among all four species that occurred approximately 50-60 million years ago (Mya); an event that probably occurred before the major radiation of the grasses. In the Solanaceae, we found evidence for a conserved duplication event approximately 50-52 Mya. A duplication in soybean occurred approximately 44 Mya and a duplication in Medicago about 58 Mya. Comparing synonymous and nonsynonymous distances allowed us to determine that most duplicate gene pairs are under purifying, negative selection. We calculated Pearson's correlation coefficients to provide us with a measure of how gene expression patterns have changed between duplicate pairs, and compared this across evolutionary distances. This analysis showed that some duplicates seemed to retain expression patterns between pairs, whereas others showed uncorrelated expression.
Finite mixture models are being used increasingly to model a wide variety of random phenomena for clustering, classification and density estimation. is a powerful and popular package which allows modelling of data as a Gaussian finite mixture with different covariance structures and different numbers of mixture components, for a variety of purposes of analysis. Recently, version 5 of the package has been made available on CRAN. This updated version adds new covariance structures, dimension reduction capabilities for visualisation, model selection criteria, initialisation strategies for the EM algorithm, and bootstrap-based inference, making it a full-featured R package for data analysis via finite mixture modelling.
Alignment quality may have as much impact on phylogenetic reconstruction as the phylogenetic methods used. Not only the alignment algorithm, but also the method used to deal with the most problematic alignment regions, may have a critical effect on the final tree. Although some authors remove such problematic regions, either manually or using automatic methods, in order to improve phylogenetic performance, others prefer to keep such regions to avoid losing any information. Our aim in the present work was to examine whether phylogenetic reconstruction improves after alignment cleaning or not. Using simulated protein alignments with gaps, we tested the relative performance in diverse phylogenetic analyses of the whole alignments versus the alignments with problematic regions removed with our previously developed Gblocks program. We also tested the performance of more or less stringent conditions in the selection of blocks. Alignments constructed with different alignment methods (ClustalW, Mafft, and Probcons) were used to estimate phylogenetic trees by maximum likelihood, neighbor joining, and parsimony. We show that, in most alignment conditions, and for alignments that are not too short, removal of blocks leads to better trees. That is, despite losing some information, there is an increase in the actual phylogenetic signal. Overall, the best trees are obtained by maximum-likelihood reconstruction of alignments cleaned by Gblocks. In general, a relaxed selection of blocks is better for short alignment, whereas a stringent selection is more adequate for longer ones. Finally, we show that cleaned alignments produce better topologies although, paradoxically, with lower bootstrap. This indicates that divergent and problematic alignment regions may lead, when present, to apparently better supported although, in fact, more biased topologies.
The phylogeny and biogeography of the genus were evaluated by using phylogenetic trees derived from analysis of nuclear ribosomal internal transcribed spacer (ITS) sequences, the plastid L intron, the L-F spacer, and the B-L spacer. This genus was divided into three subgenera:,, and. Each of them is monophyletic with high bootstrap supports according to the highly resolved phylogenetic tree reconstructed by combined sequences. There are five sections within the subgenus, including,,,, and. The subgenus is phylogenetic basal, which suggesting that is comprising more ancestral characters than other subgenera. The evolutionary trend of genus was deduced based on the maximum likelihood (ML) tree and Bayesian Evolutionary Analysis Sampling Trees (BEAST). Reconstruct Ancestral State in Phylogenies (RASP) analyses based on the combined sequence data. The biogeographic analysis indicates that species were firstly derived in Southern China and Southeast Asia, subsequently dispersed into the Southeast Asian archipelagoes. The subgenera was likely derived after these historical dispersals and vicariance events. Our research reveals the relevance of the differentiation of in Southeast Asia and geological history. Moreover, the biogeographic analysis explains that the significant evolutionary hotspots of these orchids in the Sundaland and Wallacea might be attributed to repeated migration and isolation events between the south-eastern Asia mainland and the Sunda Super Islands.Copyright © 2020 Tsai, Liao, Ko, Chen and Chiang.
Stress acclimating plants respond to abiotic and biotic stress by remodeling membrane fluidity and by releasing alpha-linolenic (18:3) from membrane lipids. The modification of membrane fluidity is mediated by changes in unsaturated fatty acid levels, a function provided in part by the regulated activity of fatty acid desaturases. Adjustment of membrane fluidity maintains an environment suitable for the function of critical integral proteins during stress. alpha-Linolenic acid, released from membrane lipid by regulated lipase activity, is the precursor molecule for phyto-oxylipin biosynthesis. The modulation of chloroplast oleic acid (18:1) levels is central to the normal expression of defense responses to pathogens in Arabidopsis. Oleic (18:1) and linolenic (18:2) acid levels, in part, regulate development, seed colonization, and mycotoxin production by Aspergillus spp.
Abiotic stress is one of the severe stresses of environment that lowers the growth and yield of any crop even on irrigated land throughout the world. A major phytohormone abscisic acid (ABA) plays an essential part in acting toward varied range of stresses like heavy metal stress, drought, thermal or heat stress, high level of salinity, low temperature, and radiation stress. Its role is also elaborated in various developmental processes including seed germination, seed dormancy, and closure of stomata. ABA acts by modifying the expression level of gene and subsequent analysis of cis- and trans-acting regulatory elements of responsive promoters. It also interacts with the signaling molecules of processes involved in stress response and development of seeds. On the whole, the stress to a plant can be susceptible or tolerant by taking into account the coordinated activities of various stress-responsive genes. Numbers of transcription factor are involved in regulating the expression of ABA responsive genes by acting together with their respective cis- acting elements. Hence, for improvement in stress-tolerance capacity of plants, it is necessary to understand the mechanism behind it. On this ground, this article enlightens the importance and role of ABA signaling with regard to various stresses as well as regulation of ABA biosynthetic pathway along with the transcription factors for stress tolerance.
Polyploidy is a prominent process in plants and has been significant in the evolutionary history of vertebrates and other eukaryotes. In plants, interdisciplinary approaches combining phylogenetic and molecular genetic perspectives have enhanced our awareness of the myriad genetic interactions made possible by polyploidy. Here, processes and mechanisms of gene and genome evolution in polyploids are reviewed. Genes duplicated by polyploidy may retain their original or similar function, undergo diversification in protein function or regulation, or one copy may become silenced through mutational or epigenetic means. Duplicated genes also may interact through inter-locus recombination, gene conversion, or concerted evolution. Recent experiments have illuminated important processes in polyploids that operate above the organizational level of duplicated genes. These include inter-genomic chromosomal exchanges, saltational, non-Mendelian genomic evolution in nascent polyploids, inter-genomic invasion, and cytonuclear stabilization. Notwithstanding many recent insights, much remains to be learned about many aspects of polyploid evolution, including: the role of transposable elements in structural and regulatory gene evolution; processes and significance of epigenetic silencing; underlying controls of chromosome pairing; mechanisms and functional significance of rapid genome changes; cytonuclear accommodation; and coordination of regulatory factors contributed by two, sometimes divergent progenitor genomes. Continued application of molecular genetic approaches to questions of polyploid genome evolution holds promise for producing lasting insight into processes by which novel genotypes are generated and ultimately into how polyploidy facilitates evolution and adaptation.
We present the 1.06 Gb sequenced genome of Gastrodia elata, an obligate mycoheterotrophic plant, which contains 18,969 protein-coding genes. Many genes conserved in other plant species have been deleted from the G. elata genome, including most of those for photosynthesis. Additional evidence of the influence of genome plasticity in the adaptation of this mycoheterotrophic lifestyle is evident in the large number of gene families that are expanded in G. elata, including glycoside hydrolases and urease that likely facilitate the digestion of hyphae are expanded, as are genes associated with strigolactone signaling, and ATPases that may contribute to the atypical energy metabolism. We also find that the plastid genome of G. elata is markedly smaller than that of green plant species while its mitochondrial genome is one of the largest observed to date. Our report establishes a foundation for studying adaptation to a mycoheterotrophic lifestyle.
Since 65 million years ago (Ma), Earth's climate has undergone a significant and complex evolution, the finer details of which are now coming to light through investigations of deep-sea sediment cores. This evolution includes gradual trends of warming and cooling driven by tectonic processes on time scales of 10(5) to 10(7) years, rhythmic or periodic cycles driven by orbital processes with 10(4)- to 10(6)-year cyclicity, and rare rapid aberrant shifts and extreme climate transients with durations of 10(3) to 10(5) years. Here, recent progress in defining the evolution of global climate over the Cenozoic Era is reviewed. We focus primarily on the periodic and anomalous components of variability over the early portion of this era, as constrained by the latest generation of deep-sea isotope records. We also consider how this improved perspective has led to the recognition of previously unforeseen mechanisms for altering climate.
Ancient whole-genome duplications (WGDs) have been uncovered in almost all major lineages of life on Earth and the search for traces or remnants of such events has become standard practice in most genome analyses. This is especially true for plants, where ancient WGDs are abundant. Common approaches to find evidence for ancient WGDs include the construction of KS distributions and the analysis of intragenomic colinearity. Despite the increased interest in WGDs and the acknowledgment of their evolutionary importance, user-friendly and comprehensive tools for their analysis are lacking. Here, we present an easy to use command-line tool for KS distribution construction named wgd. The wgd suite provides commonly used KS and colinearity analysis workflows together with tools for modeling and visualization, rendering these analyses accessible to genomics researchers in a convenient manner.wgd is free and open source software implemented in Python and is available at https://github.com/arzwa/wgd.Supplementary data are available at Bioinformatics online.© The Author(s) 2018. Published by Oxford University Press.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}