
植物学报 ›› 2015, Vol. 50 ›› Issue (4): 460-472.DOI: 10.11983/CBB14144 cstr: 32102.14.CBB14144
严玫, 张新友*( ), 韩锁义, 黄冰艳, 董文召, 刘华, 孙子淇, 张忠信, 汤丰收
), 韩锁义, 黄冰艳, 董文召, 刘华, 孙子淇, 张忠信, 汤丰收
收稿日期:2014-08-06
接受日期:2015-01-27
出版日期:2015-07-01
发布日期:2015-05-07
通讯作者:
张新友
作者简介:? 共同第一作者
基金资助:Mei Yan, Xinyou Zhang*, Suoyi Han, Bingyan Huang, Wenzhao Dong, Hua Liu, Ziqi Sun
Received:2014-08-06
Accepted:2015-01-27
Online:2015-07-01
Published:2015-05-07
Contact:
Zhang Xinyou
About author:? These authors contributed equally to this paper
摘要: 通过关联分析法发掘与花生(Arachis hypogaea)产量性状显著关联、同时又在花生基因组上随机分布的SSR位点及优异等位变异, 可了解产量相关基因区域的分布特点, 有助于利用分子标记辅助选择方法选育高产花生新品种。选用64个SSR标记, 采用MLM (Q+K)方法对166份花生资源进行全基因组关联分析。结果表明, 通过聚类分析和结构划分, 供试群体受其综合性状遗传特点和来源地域的影响可被划分成7个亚群, 聚类结果与群体结构基本一致, 同时群体特点与材料来源地的生态划分符合同类聚集的规律。通过对6个产量相关性状的3年数值的关联分析, 分别发掘出SSR位点有20个、33个和26个, 2年以上重复检出的SSR位点有13个(P<0.05), 各SSR位点的表型变异解释率范围为0.011-0.348 1, 平均为0.067 3; 共检出590个等位变异, 平均每个标记位点12.29个, 表型变异解释率值最高的是与单株果数呈显著关联的位点TC1A02 (P<0.001), 含21个等位变异; 与产量构成主要因子紧密关联的位点中, 百果重的TC1A02-C470 (+41.588 5) 、TC1A02-C560 (+40.926 1)和pPGPseq2E6-B473 (+63.953 4), 单株果数的TC1A02-C500 (+7.374 4), 单株饱果数的GM1843-E157 (+4.316 6), 可用于产量性状的分子辅助育种。
严玫, 张新友, 韩锁义, 黄冰艳, 董文召, 刘华, 孙子淇, 张忠信, 汤丰收. 花生重要农艺及产量性状的全基因组关联分析. 植物学报, 2015, 50(4): 460-472.
Mei Yan, Xinyou Zhang, Suoyi Han, Bingyan Huang, Wenzhao Dong, Hua Liu, Ziqi Sun. Genome-wide Association Study of Agronomic and Yield Traits in a Worldwide Collection of Peanut (Arachis hypogaea) Germplasm. Chinese Bulletin of Botany, 2015, 50(4): 460-472.
| SSR locus | Linkage | Position | Allele No. | Gene diversity | PIC | SSR locus | Linkage | Position | Allele No. | Gene diversity | PIC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ARS335 | LK1 | 0 | 9 | 0.824 | 0.801 | pPGPseq5D5 | LK14 | 0 | 26 | 0.911 | 0.904 |
| ARS303 | LK1 | 2 | 14 | 0.789 | 0.763 | TC2G05 | LK14 | 16 | 10 | 0.858 | 0.842 |
| ARS166 | LK1 | 6.3 | 11 | 0.835 | 0.815 | TC6E01 | LK14 | 68.8 | 28 | 0.944 | 0.941 |
| ARS535 | LK1 | 21.7 | 5 | 0.716 | 0.681 | TC2B09 | LK14 | 112.1 | 7 | 0.713 | 0.666 |
| ARS415 | LK1 | 24.4 | 8 | 0.833 | 0.811 | GM28 | LK14 | 158.8 | 10 | 0.765 | 0.733 |
| GC-94 | LK1 | 28.8 | 14 | 0.758 | 0.737 | ARS203 | LK15 | 30.2 | 17 | 0.812 | 0.788 |
| GC-104 | LK1 | 44.4 | 15 | 0.868 | 0.853 | TC1A02 | LK15 | 52.2 | 21 | 0.764 | 0.734 |
| pPGSseq19G7 | LK2 | 0 | 8 | 0.84 | 0.82 | PM54 | LK16 | 0 | 10 | 0.796 | 0.765 |
| PM81 | LK2 | 10.6 | 6 | 0.36 | 0.338 | pPGPseq3E10 | LK17 | 0 | 9 | 0.814 | 0.787 |
| ARS392 | LK2 | 51.6 | 10 | 0.815 | 0.788 | pPGPseq4H11 | LK17 | 13.4 | 15 | 0.731 | 0.697 |
| ARS251 | LK2 | 60.3 | 12 | 0.862 | 0.848 | PMc588 | - | 6 | 0.635 | 0.569 | |
| ARS180 | LK2 | 73 | 12 | 0.874 | 0.86 | pPGSseq15D3 | - | 10 | 0.724 | 0.688 | |
| GM553 | LK3 | 0 | 12 | 0.847 | 0.829 | pPGSseq18G1 | - | 6 | 0.637 | 0.571 | |
| pPGSseq16F1 | LK3 | 36.3 | 6 | 0.799 | 0.768 | pPGPseq2E6 | - | 19 | 0.812 | 0.792 | |
| ARS173 | LK4 | 0 | 16 | 0.844 | 0.825 | pPGPseq2D12B | - | 7 | 0.711 | 0.678 | |
| ARS298 | LK4 | 0.891 | 19 | 0.874 | 0.861 | PM137 | - | 13 | 0.809 | 0.786 | |
| GM1843 | LK4 | 3.464 | 9 | 0.778 | 0.744 | PM377 | - | 13 | 0.798 | 0.775 | |
| GM401 | LK4 | 51.1 | 9 | 0.744 | 0.699 | TC4G02 | - | 20 | 0.837 | 0.823 | |
| ARS313 | LK6 | 25.4 | 5 | 0.448 | 0.4 | GM529 | - | 9 | 0.744 | 0.704 | |
| EM-87 | LK7 | 4.8 | 18 | 0.829 | 0.808 | TC1G04 | - | 10 | 0.674 | 0.626 | |
| ARS376 | LK7 | 25.2 | 13 | 0.839 | 0.82 | TC2E05 | - | 13 | 0.719 | 0.675 | |
| pPGSseq17E3 | LK7 | 49.9 | 6 | 0.705 | 0.661 | TC3G05 | - | 11 | 0.697 | 0.646 | |
| ARS644 | LK7 | 62.6 | 4 | 0.534 | 0.449 | TC4G02 | - | 11 | 0.823 | 0.801 | |
| pPGPseq1B9 | LK8 | 0 | 11 | 0.816 | 0.791 | RN26G09 | - | 14 | 0.89 | 0.879 | |
| ARS545 | LK9 | 33.9 | 15 | 0.867 | 0.852 | RN34A10 | - | 9 | 0.69 | 0.635 | |
| ARS127 | LK10 | 33.3 | 3 | 0.549 | 0.451 | ARS98 | - | 10 | 0.818 | 0.794 | |
| ARS357 | LK11 | 6.5 | 7 | 0.823 | 0.799 | ARS108 | - | 14 | 0.869 | 0.856 | |
| PM436 | LK12 | 41.7 | 9 | 0.822 | 0.8 | ARS124 | - | 7 | 0.75 | 0.706 | |
| pPGSseq14H6 | LK12 | 47.7 | 11 | 0.829 | 0.808 | ARS318 | - | 13 | 0.88 | 0.868 | |
| pPGSseq15C12 | LK13 | 0 | 17 | 0.855 | 0.841 | ARS338 | - | 16 | 0.859 | 0.844 | |
| pPGSseq17F6 | LK13 | 50.8 | 21 | 0.917 | 0.911 | ARS369 | - | 9 | 0.615 | 0.567 | |
| ARS606 | - | 10 | 0.808 | 0.78 | |||||||
| GM384 | - | 6 | 0.751 | 0.711 | |||||||
| Mean | 11.6 | 0.777 | 0.748 |
表1 SSR标记的多态性
Table 1 Polymorphism information of SSR
| SSR locus | Linkage | Position | Allele No. | Gene diversity | PIC | SSR locus | Linkage | Position | Allele No. | Gene diversity | PIC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ARS335 | LK1 | 0 | 9 | 0.824 | 0.801 | pPGPseq5D5 | LK14 | 0 | 26 | 0.911 | 0.904 |
| ARS303 | LK1 | 2 | 14 | 0.789 | 0.763 | TC2G05 | LK14 | 16 | 10 | 0.858 | 0.842 |
| ARS166 | LK1 | 6.3 | 11 | 0.835 | 0.815 | TC6E01 | LK14 | 68.8 | 28 | 0.944 | 0.941 |
| ARS535 | LK1 | 21.7 | 5 | 0.716 | 0.681 | TC2B09 | LK14 | 112.1 | 7 | 0.713 | 0.666 |
| ARS415 | LK1 | 24.4 | 8 | 0.833 | 0.811 | GM28 | LK14 | 158.8 | 10 | 0.765 | 0.733 |
| GC-94 | LK1 | 28.8 | 14 | 0.758 | 0.737 | ARS203 | LK15 | 30.2 | 17 | 0.812 | 0.788 |
| GC-104 | LK1 | 44.4 | 15 | 0.868 | 0.853 | TC1A02 | LK15 | 52.2 | 21 | 0.764 | 0.734 |
| pPGSseq19G7 | LK2 | 0 | 8 | 0.84 | 0.82 | PM54 | LK16 | 0 | 10 | 0.796 | 0.765 |
| PM81 | LK2 | 10.6 | 6 | 0.36 | 0.338 | pPGPseq3E10 | LK17 | 0 | 9 | 0.814 | 0.787 |
| ARS392 | LK2 | 51.6 | 10 | 0.815 | 0.788 | pPGPseq4H11 | LK17 | 13.4 | 15 | 0.731 | 0.697 |
| ARS251 | LK2 | 60.3 | 12 | 0.862 | 0.848 | PMc588 | - | 6 | 0.635 | 0.569 | |
| ARS180 | LK2 | 73 | 12 | 0.874 | 0.86 | pPGSseq15D3 | - | 10 | 0.724 | 0.688 | |
| GM553 | LK3 | 0 | 12 | 0.847 | 0.829 | pPGSseq18G1 | - | 6 | 0.637 | 0.571 | |
| pPGSseq16F1 | LK3 | 36.3 | 6 | 0.799 | 0.768 | pPGPseq2E6 | - | 19 | 0.812 | 0.792 | |
| ARS173 | LK4 | 0 | 16 | 0.844 | 0.825 | pPGPseq2D12B | - | 7 | 0.711 | 0.678 | |
| ARS298 | LK4 | 0.891 | 19 | 0.874 | 0.861 | PM137 | - | 13 | 0.809 | 0.786 | |
| GM1843 | LK4 | 3.464 | 9 | 0.778 | 0.744 | PM377 | - | 13 | 0.798 | 0.775 | |
| GM401 | LK4 | 51.1 | 9 | 0.744 | 0.699 | TC4G02 | - | 20 | 0.837 | 0.823 | |
| ARS313 | LK6 | 25.4 | 5 | 0.448 | 0.4 | GM529 | - | 9 | 0.744 | 0.704 | |
| EM-87 | LK7 | 4.8 | 18 | 0.829 | 0.808 | TC1G04 | - | 10 | 0.674 | 0.626 | |
| ARS376 | LK7 | 25.2 | 13 | 0.839 | 0.82 | TC2E05 | - | 13 | 0.719 | 0.675 | |
| pPGSseq17E3 | LK7 | 49.9 | 6 | 0.705 | 0.661 | TC3G05 | - | 11 | 0.697 | 0.646 | |
| ARS644 | LK7 | 62.6 | 4 | 0.534 | 0.449 | TC4G02 | - | 11 | 0.823 | 0.801 | |
| pPGPseq1B9 | LK8 | 0 | 11 | 0.816 | 0.791 | RN26G09 | - | 14 | 0.89 | 0.879 | |
| ARS545 | LK9 | 33.9 | 15 | 0.867 | 0.852 | RN34A10 | - | 9 | 0.69 | 0.635 | |
| ARS127 | LK10 | 33.3 | 3 | 0.549 | 0.451 | ARS98 | - | 10 | 0.818 | 0.794 | |
| ARS357 | LK11 | 6.5 | 7 | 0.823 | 0.799 | ARS108 | - | 14 | 0.869 | 0.856 | |
| PM436 | LK12 | 41.7 | 9 | 0.822 | 0.8 | ARS124 | - | 7 | 0.75 | 0.706 | |
| pPGSseq14H6 | LK12 | 47.7 | 11 | 0.829 | 0.808 | ARS318 | - | 13 | 0.88 | 0.868 | |
| pPGSseq15C12 | LK13 | 0 | 17 | 0.855 | 0.841 | ARS338 | - | 16 | 0.859 | 0.844 | |
| pPGSseq17F6 | LK13 | 50.8 | 21 | 0.917 | 0.911 | ARS369 | - | 9 | 0.615 | 0.567 | |
| ARS606 | - | 10 | 0.808 | 0.78 | |||||||
| GM384 | - | 6 | 0.751 | 0.711 | |||||||
| Mean | 11.6 | 0.777 | 0.748 |
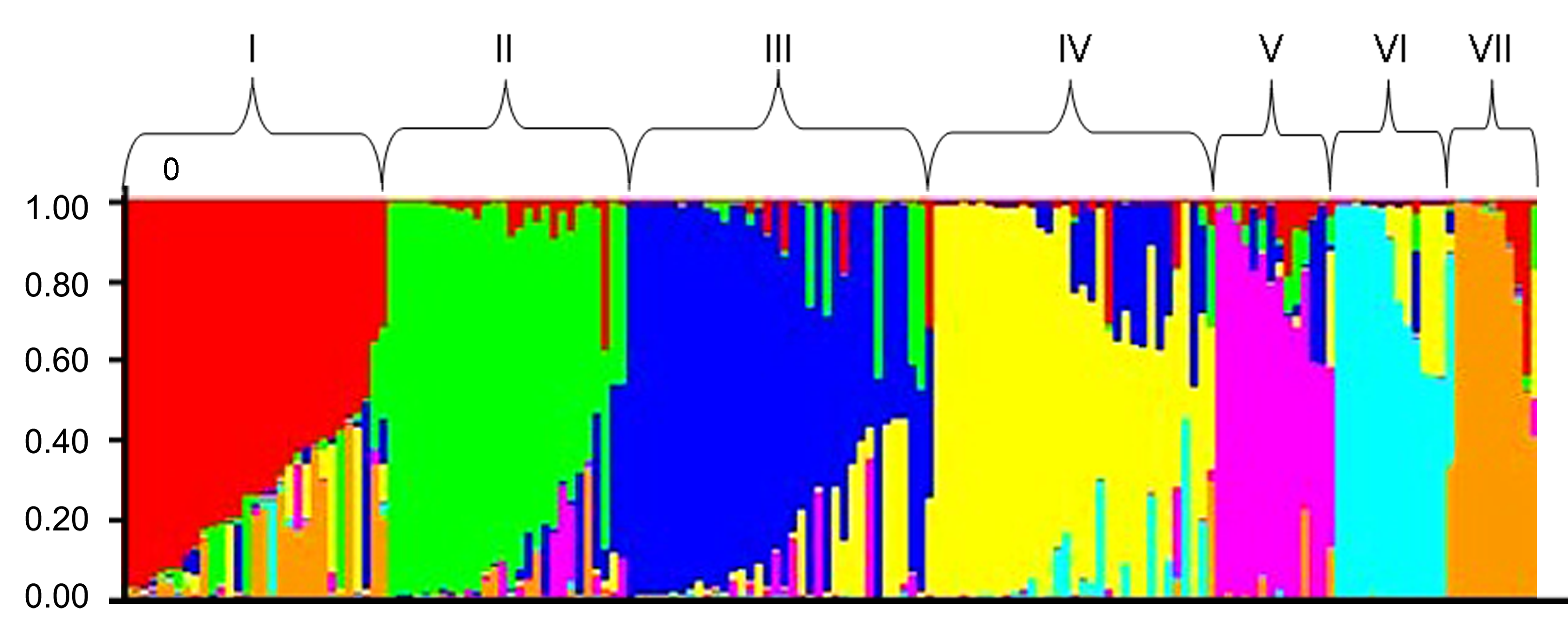
图2 166份花生种质的群体结构分析
Figure 2 Population structure of 166 peanut germplasm
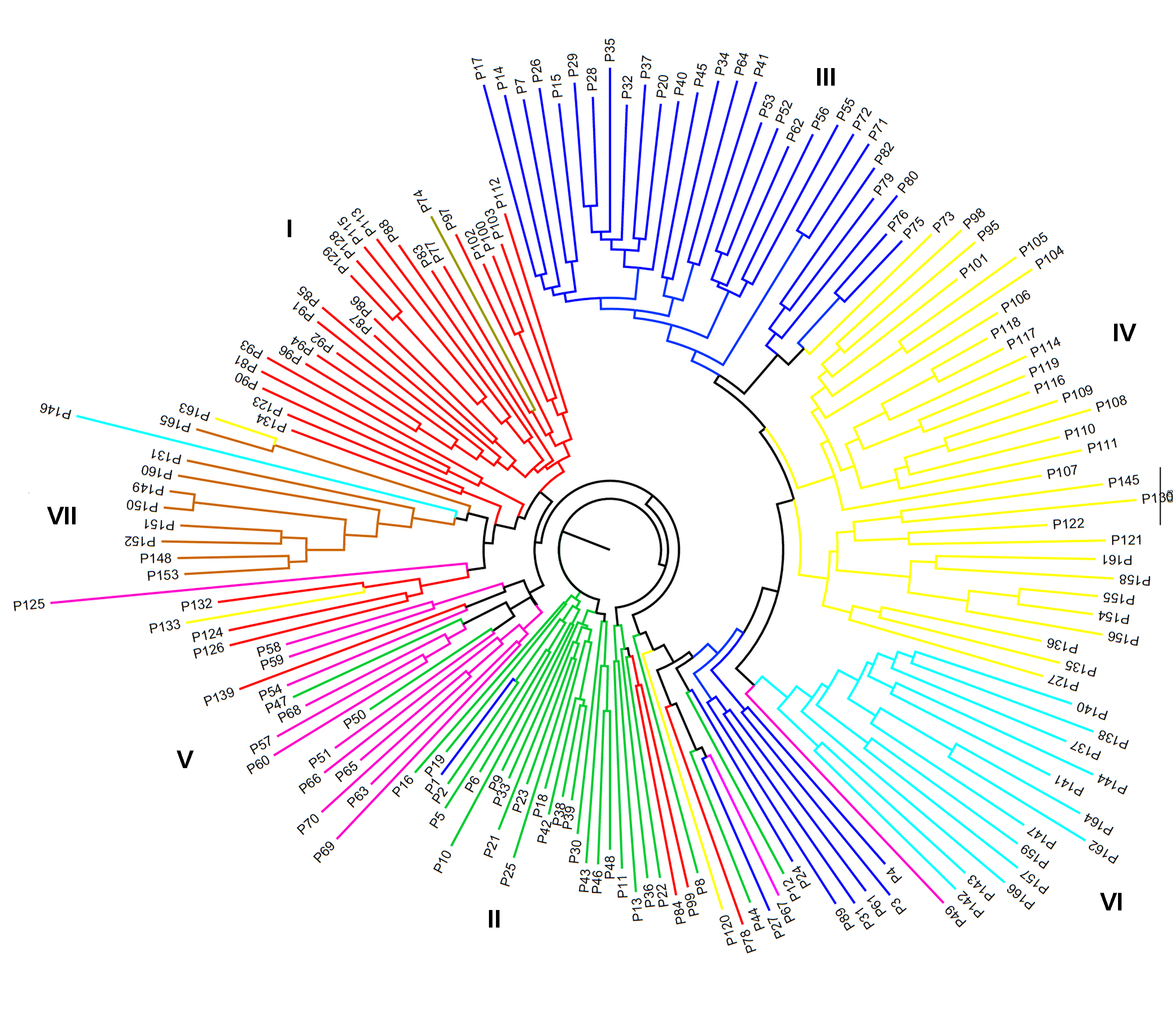
图3 166份花生种质基于Nei’s 1983聚类/邻接法构建的系统发育树
Figure 3 The dendrogram of the 166 peanut germplasm based on Nei’s 1983/Neighbor-joining cluster
| Geographic eco-type | Structure | Total | χ2 test | ||||||
|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | |||
| Southeast of China | 1 | 25 | 18 | 2 | 46 | ||||
| Huang River region of China | 17 | 23 | 6 | 2 | 7 | 1 | 56 | χ2=166** | |
| Yangtzi River region of China | 11 | 6 | 4 | 3 | 6 | 1 | 31 | P<0.000 1 | |
| Yunnan and Guizhou Plateau | 1 | 2 | 3 | ||||||
| World wide origin | 1 | 8 | 13 | 8 | 30 | χ20.01, 24= 42.98 | |||
| Total | 31 | 29 | 35 | 33 | 15 | 13 | 10 | 166 | |
表2 166份花生种质SSR标记模型聚类与来源地生态划分的相关性
Table 2 Association between model-based clusters and geographic eco-types of 166 peanut germplasm
| Geographic eco-type | Structure | Total | χ2 test | ||||||
|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | |||
| Southeast of China | 1 | 25 | 18 | 2 | 46 | ||||
| Huang River region of China | 17 | 23 | 6 | 2 | 7 | 1 | 56 | χ2=166** | |
| Yangtzi River region of China | 11 | 6 | 4 | 3 | 6 | 1 | 31 | P<0.000 1 | |
| Yunnan and Guizhou Plateau | 1 | 2 | 3 | ||||||
| World wide origin | 1 | 8 | 13 | 8 | 30 | χ20.01, 24= 42.98 | |||
| Total | 31 | 29 | 35 | 33 | 15 | 13 | 10 | 166 | |
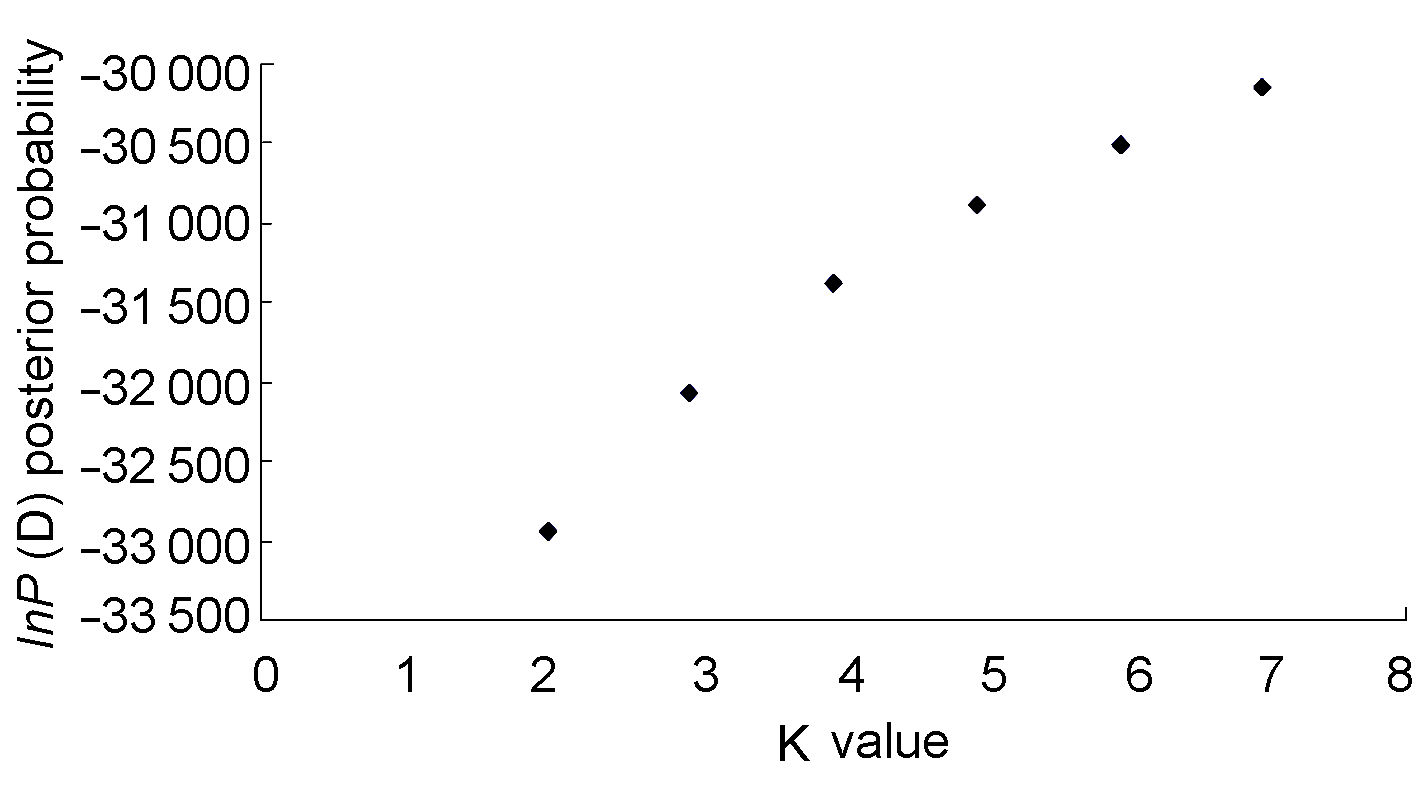
图1 不同亚群K值的后验概率lnP(D)的变异趋势
Figure 1 The trendline of posterior probabilities (lnP(D)) for each hypothetic number of subpopulations (K)
| Trait | I | II | III | IV | V | VI | VII |
|---|---|---|---|---|---|---|---|
| 1 | 45.21±1.92 | 46.93±0.83 | 48.82±1.25 | 55.92±1.97 | 45.58±1.59 | 79.51±2.81 | 53.80±2.89 |
| 2 | 53.77±1.24 | 55.93±1.18 | 58.96±1.42 | 66.95±2.23 | 54.14±2.06 | 95.07±3.64 | 70.63±3.07 |
| 3 | 11.37±0.50 | 11.55±0.60 | 10.31±0.32 | 10.33±0.36 | 10.74±0.66 | 10.38±0.88 | 19.02±1.40 |
| 4 | 8.99±0.16 | 8.96±0.24 | 7.29±0.15 | 7.37±0.20 | 8.57±0.40 | 6.88±0.32 | 11.1±0.67 |
| 5 | 28.64±0.79 | 27.91±0.61 | 21.20±0.92 | 20.80±1.01 | 27.3±1.41 | 15.74±1.12 | 19.45±1.85 |
| 6 | 23.71±0.52 | 22.51±0.48 | 20.08±0.50 | 21.68±0.75 | 24±0.83 | 17.89±0.89 | 22.56±0.86 |
| 7 | 16.8±0.36 | 15.3±0.46 | 14.6±0.41 | 16.53±0.54 | 16.01±0.69 | 12.27±0.70 | 13.26±0.76 |
| 8 | 176.58±6.05 | 176.93±3.93 | 139.16±3.47 | 135.32±6.46 | 172.81±6.34 | 120.17±6.99 | 123.78±11.54 |
| 9 | 75.47±2.33 | 76.93±1.59 | 54.92±1.54 | 53.59±2.45 | 69.60±2.78 | 39.85±2.34 | 55.05±3.82 |
| 10 | 0.51±0.01 | 0.51±0.01 | 0.48±0.01 | 0.49±0.01 | 0.51±0.01 | 0.49±0.01 | 0.46±0.02 |
表3 依据群体结构划分的各亚群内品种的农艺与产量性状比对(3年平均值±标准误)
Table 3 The comparison of agronomic and yield traits between 7 subgroups by means±SD of 3 years
| Trait | I | II | III | IV | V | VI | VII |
|---|---|---|---|---|---|---|---|
| 1 | 45.21±1.92 | 46.93±0.83 | 48.82±1.25 | 55.92±1.97 | 45.58±1.59 | 79.51±2.81 | 53.80±2.89 |
| 2 | 53.77±1.24 | 55.93±1.18 | 58.96±1.42 | 66.95±2.23 | 54.14±2.06 | 95.07±3.64 | 70.63±3.07 |
| 3 | 11.37±0.50 | 11.55±0.60 | 10.31±0.32 | 10.33±0.36 | 10.74±0.66 | 10.38±0.88 | 19.02±1.40 |
| 4 | 8.99±0.16 | 8.96±0.24 | 7.29±0.15 | 7.37±0.20 | 8.57±0.40 | 6.88±0.32 | 11.1±0.67 |
| 5 | 28.64±0.79 | 27.91±0.61 | 21.20±0.92 | 20.80±1.01 | 27.3±1.41 | 15.74±1.12 | 19.45±1.85 |
| 6 | 23.71±0.52 | 22.51±0.48 | 20.08±0.50 | 21.68±0.75 | 24±0.83 | 17.89±0.89 | 22.56±0.86 |
| 7 | 16.8±0.36 | 15.3±0.46 | 14.6±0.41 | 16.53±0.54 | 16.01±0.69 | 12.27±0.70 | 13.26±0.76 |
| 8 | 176.58±6.05 | 176.93±3.93 | 139.16±3.47 | 135.32±6.46 | 172.81±6.34 | 120.17±6.99 | 123.78±11.54 |
| 9 | 75.47±2.33 | 76.93±1.59 | 54.92±1.54 | 53.59±2.45 | 69.60±2.78 | 39.85±2.34 | 55.05±3.82 |
| 10 | 0.51±0.01 | 0.51±0.01 | 0.48±0.01 | 0.49±0.01 | 0.51±0.01 | 0.49±0.01 | 0.46±0.02 |
| Trait | Means±SD | Range | CV (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2011 | 2012 | 2013 | 2011 | 2012 | 2013 | 2011 | 2012 | 2013 | |||
| 1 | 43.73±10.48 | 56.08±12.42 | 55.17±15.24 | 24.60-77.80 | 33.00-102.99 | 33.25-116.70 | 10.48 | 22.16 | 27.63 | ||
| 2 | 55.33±12.76 | 64.81±14.06 | 66.29±18.11 | 32.00-100.60 | 38.00-130.33 | 36.48-140.6 | 12.76 | 21.69 | 27.32 | ||
| 3 | 12.00±3.68 | 10.46±3.41 | 11.48±3.73 | 4.89-25.15 | 5.10-24.00 | 5.30-31.60 | 3.68 | 32.58 | 32.47 | ||
| 4 | 8.27±1.51 | 8.30±2.01 | 8.05±1.99 | 4.50-15.80 | 4.92-15.90 | 3.82-14.6 | 1.51 | 24.23 | 24.57 | ||
| 5 | 19.75±7.76 | 25.76±5.87 | 25.61±8.48 | 3.40-36.5 | 10.00-40.85 | 3.04-56.00 | 7.76 | 22.78 | 33.13 | ||
| 6 | 19.37±5.57 | 25.44±4.99 | 20.69±4.99 | 7.05-31.90 | 12.93-57.62 | 6.21-39.70 | 5.57 | 19.59 | 24.13 | ||
| 7 | 11.89±4.76 | 20.01±3.5 | 14.25±4.07 | 3.60-28.10 | 10.40-32.13 | 3.09-35.20 | 4.76 | 17.49 | 28.58 | ||
| 8 | 129.93±37.76 | 146.84±31.35 | 181.07±45.87 | 44.40-240.00 | 49.00-228.28 | 62.00-298.00 | 37.76 | 21.35 | 25.34 | ||
| 9 | 56.73±17.25 | 57.35±14.39 | 73.39±20.73 | 10..0-120.85 | 17.00-91.50 | 27.50-127.50 | 17.25 | 25.10 | 28.25 | ||
| 10 | 0.53±0.09 | 0.70±0.06 | 0.46±0.07 | 0.27-0.72 | 0.35-0.82 | 0.11-0.62 | 0.09 | 8.96 | 26.65 | ||
表4 花生种质在2011-2013年主要农艺和产量性状的变异分析
Table 4 Variation analysis of agronomic and yield traits among peanut germplasm in 2011-2013
| Trait | Means±SD | Range | CV (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2011 | 2012 | 2013 | 2011 | 2012 | 2013 | 2011 | 2012 | 2013 | |||
| 1 | 43.73±10.48 | 56.08±12.42 | 55.17±15.24 | 24.60-77.80 | 33.00-102.99 | 33.25-116.70 | 10.48 | 22.16 | 27.63 | ||
| 2 | 55.33±12.76 | 64.81±14.06 | 66.29±18.11 | 32.00-100.60 | 38.00-130.33 | 36.48-140.6 | 12.76 | 21.69 | 27.32 | ||
| 3 | 12.00±3.68 | 10.46±3.41 | 11.48±3.73 | 4.89-25.15 | 5.10-24.00 | 5.30-31.60 | 3.68 | 32.58 | 32.47 | ||
| 4 | 8.27±1.51 | 8.30±2.01 | 8.05±1.99 | 4.50-15.80 | 4.92-15.90 | 3.82-14.6 | 1.51 | 24.23 | 24.57 | ||
| 5 | 19.75±7.76 | 25.76±5.87 | 25.61±8.48 | 3.40-36.5 | 10.00-40.85 | 3.04-56.00 | 7.76 | 22.78 | 33.13 | ||
| 6 | 19.37±5.57 | 25.44±4.99 | 20.69±4.99 | 7.05-31.90 | 12.93-57.62 | 6.21-39.70 | 5.57 | 19.59 | 24.13 | ||
| 7 | 11.89±4.76 | 20.01±3.5 | 14.25±4.07 | 3.60-28.10 | 10.40-32.13 | 3.09-35.20 | 4.76 | 17.49 | 28.58 | ||
| 8 | 129.93±37.76 | 146.84±31.35 | 181.07±45.87 | 44.40-240.00 | 49.00-228.28 | 62.00-298.00 | 37.76 | 21.35 | 25.34 | ||
| 9 | 56.73±17.25 | 57.35±14.39 | 73.39±20.73 | 10..0-120.85 | 17.00-91.50 | 27.50-127.50 | 17.25 | 25.10 | 28.25 | ||
| 10 | 0.53±0.09 | 0.70±0.06 | 0.46±0.07 | 0.27-0.72 | 0.35-0.82 | 0.11-0.62 | 0.09 | 8.96 | 26.65 | ||
| Trait | MH | LB | TB | PWP | NB | PN | NW | PW | SW |
|---|---|---|---|---|---|---|---|---|---|
| LB | 0.953** | 1 | |||||||
| TB | -0.094 | -0.002 | 1 | ||||||
| PWP | -0.476** | -0.526** | -0.025 | 1 | |||||
| NB | -0.324** | -0.272** | 0.794** | 0.410** | 1 | ||||
| PN | -0.216** | -0.220** | 0.193* | 0.573** | 0.443** | 1 | |||
| NW | -0.266** | -0.309** | -0.042 | 0.593** | 0.205** | 0.765** | 1 | ||
| PW | -0.432** | -0.478** | -0.108 | 0.786** | 0.267** | 0.118 | 0.189* | 1 | |
| SW | -0.539** | -0.569** | 0.040 | 0.755** | 0.416** | 0.183* | 0.165* | 0.901** | 1 |
| SP | -0.151 | -0.129 | -0.100 | -0.106 | -0.027 | -0.185* | -0.238** | 0.106 | 0.257** |
表5 166份花生种质主要农艺性状与产量性状的相关分析
Table 5 The correlation coefficient of agronomic and yield traits in 166 peanut germplasm
| Trait | MH | LB | TB | PWP | NB | PN | NW | PW | SW |
|---|---|---|---|---|---|---|---|---|---|
| LB | 0.953** | 1 | |||||||
| TB | -0.094 | -0.002 | 1 | ||||||
| PWP | -0.476** | -0.526** | -0.025 | 1 | |||||
| NB | -0.324** | -0.272** | 0.794** | 0.410** | 1 | ||||
| PN | -0.216** | -0.220** | 0.193* | 0.573** | 0.443** | 1 | |||
| NW | -0.266** | -0.309** | -0.042 | 0.593** | 0.205** | 0.765** | 1 | ||
| PW | -0.432** | -0.478** | -0.108 | 0.786** | 0.267** | 0.118 | 0.189* | 1 | |
| SW | -0.539** | -0.569** | 0.040 | 0.755** | 0.416** | 0.183* | 0.165* | 0.901** | 1 |
| SP | -0.151 | -0.129 | -0.100 | -0.106 | -0.027 | -0.185* | -0.238** | 0.106 | 0.257** |
| Trait | SSR locus | MLM-model R2 | P value | |||||
|---|---|---|---|---|---|---|---|---|
| 2011 | 2012 | 2013 | 2011 | 2012 | 2013 | |||
| Number of pod-bearing branches | pPGSseq15D3 | 0.065 3 | 0.067 7 | 0.009 5 | 0.017 1 | |||
| ARS376 | 0.060 2 | 0.048 9 | 0.016 5 | 0.030 1 | ||||
| ARS535 | 0.038 1 | 0.026 6 | 0.003 7 | 0.023 6 | ||||
| Pod weight per plant | PM377 | 0.046 3 | 0.057 6 | 0.023 | 0.025 4 | |||
| TC3G05 | 0.054 4 | 0.096 1 | 0.046 5 | 1.45×10-5 | ||||
| Pods number per plant | GM529 | 0.084 3 | 0.033 4 | 0.004 1 | 0.044 8 | |||
| TC1A02 | 0.348 1 | 0.179 5 | 0.014 2 | 1.42×10-10 | 1.97×10-4 | |||
| GM1843 | 0.053 7 | 0.042 4 | 0.003 5 | 0.028 2 | ||||
| Number of well filled pods per plant | PM54 | 0.051 5 | 0.077 6 | 0.011 9 | 0.015 2 | |||
| GM529 | 0.063 2 | 0.029 8 | 0.011 4 | 0.036 9 | ||||
| TC1A02 | 0.137 6 | 0.222 9 | 0.017 7 | 5.11×10-5 | ||||
| GM1843 | 0.022 4 | 0.049 4 | 0.039 7 | 0.008 5 | ||||
| 100-pod weight | PM54 | 0.039 7 | 0.045 7 | 0.055 3 | 0.037 | 0.018 6 | 5.73×10-4 | |
| pPGPseq2E6 | 0.058 3 | 0.060 9 | 0.045 1 | 0.010 7 | 3.75×10-4 | 0.007 2 | ||
| TC4G02 | 0.091 2 | 0.100 4 | 0.060 9 | 0.001 3 | 0.007 1 | 0.02 | ||
| EM-87 | 0.112 1 | 0.061 4 | 1.80×10-5 | 0.002 | ||||
| TC1A02 | 0.065 8 | 0.060 3 | 0.039 2 | 0.022 9 | ||||
| 100-seed weight | pPGSseq14H6 | 0.033 7 | 0.025 3 | 0.013 8 | 0.006 2 | 0.016 8 | ||
| EM-87 | 0.047 2 | 0.052 4 | 0.027 2 | 0.005 1 | ||||
| ARS392 | 0.037 4 | 0.036 8 | 0.022 7 | 8.60×10-4 | 0.002 7 | 0.023 9 | ||
| ARS535 | 0.014 | 0.011 | 0.022 6 | 0.035 4 | ||||
表6 采用MLM (K+Q)模型检测的连续2年以上与6个产量相关性状关联的SSR位点
Table 6 SSR associated with 6 yield related traits could be detected repeatedly over 2 years with MLM (K+Q) model
| Trait | SSR locus | MLM-model R2 | P value | |||||
|---|---|---|---|---|---|---|---|---|
| 2011 | 2012 | 2013 | 2011 | 2012 | 2013 | |||
| Number of pod-bearing branches | pPGSseq15D3 | 0.065 3 | 0.067 7 | 0.009 5 | 0.017 1 | |||
| ARS376 | 0.060 2 | 0.048 9 | 0.016 5 | 0.030 1 | ||||
| ARS535 | 0.038 1 | 0.026 6 | 0.003 7 | 0.023 6 | ||||
| Pod weight per plant | PM377 | 0.046 3 | 0.057 6 | 0.023 | 0.025 4 | |||
| TC3G05 | 0.054 4 | 0.096 1 | 0.046 5 | 1.45×10-5 | ||||
| Pods number per plant | GM529 | 0.084 3 | 0.033 4 | 0.004 1 | 0.044 8 | |||
| TC1A02 | 0.348 1 | 0.179 5 | 0.014 2 | 1.42×10-10 | 1.97×10-4 | |||
| GM1843 | 0.053 7 | 0.042 4 | 0.003 5 | 0.028 2 | ||||
| Number of well filled pods per plant | PM54 | 0.051 5 | 0.077 6 | 0.011 9 | 0.015 2 | |||
| GM529 | 0.063 2 | 0.029 8 | 0.011 4 | 0.036 9 | ||||
| TC1A02 | 0.137 6 | 0.222 9 | 0.017 7 | 5.11×10-5 | ||||
| GM1843 | 0.022 4 | 0.049 4 | 0.039 7 | 0.008 5 | ||||
| 100-pod weight | PM54 | 0.039 7 | 0.045 7 | 0.055 3 | 0.037 | 0.018 6 | 5.73×10-4 | |
| pPGPseq2E6 | 0.058 3 | 0.060 9 | 0.045 1 | 0.010 7 | 3.75×10-4 | 0.007 2 | ||
| TC4G02 | 0.091 2 | 0.100 4 | 0.060 9 | 0.001 3 | 0.007 1 | 0.02 | ||
| EM-87 | 0.112 1 | 0.061 4 | 1.80×10-5 | 0.002 | ||||
| TC1A02 | 0.065 8 | 0.060 3 | 0.039 2 | 0.022 9 | ||||
| 100-seed weight | pPGSseq14H6 | 0.033 7 | 0.025 3 | 0.013 8 | 0.006 2 | 0.016 8 | ||
| EM-87 | 0.047 2 | 0.052 4 | 0.027 2 | 0.005 1 | ||||
| ARS392 | 0.037 4 | 0.036 8 | 0.022 7 | 8.60×10-4 | 0.002 7 | 0.023 9 | ||
| ARS535 | 0.014 | 0.011 | 0.022 6 | 0.035 4 | ||||
| 100-pod weight (g) | Pods number per plant | Full pods number per plant | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SSR locus | Alleles | AI | SSR locus | Alleles | AI | SSR locus | Alleles | AI | |||
| PM54 | A113 | -22.870 6 | GM529 | B160 | -0.598 | PM54 | A113 | 1.529 2 | |||
| A181 | 34.132 4 | B219 | 0.266 2 | A181 | 0.287 7 | ||||||
| A185 | -9.174 7 | B233 | -12.889 | A185 | -0.888 4 | ||||||
| A194 | -10.121 9 | B254 | -6.598 | A194 | 0.708 7 | ||||||
| A114 | -32.339 4 | B175 | 1.505 | A114 | -2.899 9 | ||||||
| A197 | -68.583 1 | TC1A02 | C475 | -8.133 | A197 | 1.774 0 | |||||
| A200 | -35.389 9 | C500 | 7.374 5 | A200 | -3.374 6 | ||||||
| A205 | -66.972 6 | C591 | -6.488 4 | A205 | -1.658 2 | ||||||
| pPGPseq2E6 | B473 | 63.953 4 | C470 | -0.347 5 | GM529 | B160 | 0.663 6 | ||||
| B416 | 0.798 2 | C485 | -1.650 3 | B219 | 0.737 1 | ||||||
| B504 | 6.721 1 | C242 | 1.548 1 | B233 | -6.400 4 | ||||||
| B540 | -57.571 8 | C565 | -1.225 | B254 | -5.111 4 | ||||||
| B608 | -11.112 1 | C575 | 2.235 7 | B175 | 0.298 0 | ||||||
| B400 | 6.932 4 | C580 | 0.586 2 | TC1A02 | C475 | -4.915 9 | |||||
| B433 | -0.231 1 | C245 | -2.753 9 | C500 | 1.399 0 | ||||||
| B450 | 19.717 0 | C565 | 1.230 3 | C591 | -0.730 2 | ||||||
| B385 | -6.731 0 | C246 | -0.511 6 | C470 | -0.229 1 | ||||||
| B465 | 7.411 0 | C560 | -0.853 8 | C485 | -1.425 9 | ||||||
| B516 | 33.917 8 | C250 | 0.504 5 | C500 | 0.330 6 | ||||||
| B460 | 15.365 3 | C260 | -4.067 7 | C242 | -1.334 4 | ||||||
| B270 | 48.274 5 | C262 | 2.209 | ||||||||
| TC1A02 | C475 | -15.280 2 | C447 | -1.908 5 | C575 | 2.100 0 | |||||
| C500 | 7.374 5 | GM1843 | E111 | -3.135 8 | C580 | -1.821 0 | |||||
| C591 | -29.501 6 | E117 | -0.238 5 | C245 | -3.147 3 | ||||||
| C470 | 41.588 5 | E157 | -2.066 7 | C565 | 2.648 1 | ||||||
| C485 | -13.043 2 | E119 | 2.327 5 | C246 | -0.807 5 | ||||||
| C500 | -51.089 | E121 | 3.294 1 | C560 | -2.600 1 | ||||||
| C242 | 38.863 2 | E132 | -2.161 2 | C250 | -2.266 5 | ||||||
| C575 | 17.545 3 | C260 | -3.740 9 | ||||||||
| C580 | 20.984 1 | C262 | -1.317 6 | ||||||||
| C245 | -39.861 6 | C447 | -1.123 5 | ||||||||
| C565 | -28.856 2 | GM1843 | E111 | -2.869 5 | |||||||
| C246 | -37.283 | E117 | -0.629 2 | ||||||||
| C560 | 40.926 2 | E157 | 4.316 6 | ||||||||
| C250 | -62.524 5 | E119 | 0.728 9 | ||||||||
| C260 | -32.857 4 | E121 | 2.357 2 | ||||||||
| C262 | -18.781 8 | E132 | -0.283 7 | ||||||||
| C447 | -21.705 4 | AAE (positive/negative) | |||||||||
| AAE (positive/negative) | AAE (positive/negative) | PM54 | 1.074 9 | -2.205 2 | |||||||
| PM54 | 34.132 4 | -35.064 6 | GM529 | 0.885 6 | -6.695 | GM529 | 0.566 2 | -5.755 9 | |||
| pPGPseq2E6 | 22.565 6 | -18.911 5 | TC1A02 | 2.241 2 | -2.713 5 | TC1A02 | 1.619 4 | -1.958 5 | |||
| TC1A02 | 27.880 3 | -31.889 4 | GM1843 | 2.810 8 | -1.900 6 | GM1843 | 2.467 6 | -1.260 8 | |||
表7 产量性状关联位点的主要等位基因表型效应
Table 7 Phenotypic effect of main alleles at SSR loci significantly associated with yield related traits
| 100-pod weight (g) | Pods number per plant | Full pods number per plant | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SSR locus | Alleles | AI | SSR locus | Alleles | AI | SSR locus | Alleles | AI | |||
| PM54 | A113 | -22.870 6 | GM529 | B160 | -0.598 | PM54 | A113 | 1.529 2 | |||
| A181 | 34.132 4 | B219 | 0.266 2 | A181 | 0.287 7 | ||||||
| A185 | -9.174 7 | B233 | -12.889 | A185 | -0.888 4 | ||||||
| A194 | -10.121 9 | B254 | -6.598 | A194 | 0.708 7 | ||||||
| A114 | -32.339 4 | B175 | 1.505 | A114 | -2.899 9 | ||||||
| A197 | -68.583 1 | TC1A02 | C475 | -8.133 | A197 | 1.774 0 | |||||
| A200 | -35.389 9 | C500 | 7.374 5 | A200 | -3.374 6 | ||||||
| A205 | -66.972 6 | C591 | -6.488 4 | A205 | -1.658 2 | ||||||
| pPGPseq2E6 | B473 | 63.953 4 | C470 | -0.347 5 | GM529 | B160 | 0.663 6 | ||||
| B416 | 0.798 2 | C485 | -1.650 3 | B219 | 0.737 1 | ||||||
| B504 | 6.721 1 | C242 | 1.548 1 | B233 | -6.400 4 | ||||||
| B540 | -57.571 8 | C565 | -1.225 | B254 | -5.111 4 | ||||||
| B608 | -11.112 1 | C575 | 2.235 7 | B175 | 0.298 0 | ||||||
| B400 | 6.932 4 | C580 | 0.586 2 | TC1A02 | C475 | -4.915 9 | |||||
| B433 | -0.231 1 | C245 | -2.753 9 | C500 | 1.399 0 | ||||||
| B450 | 19.717 0 | C565 | 1.230 3 | C591 | -0.730 2 | ||||||
| B385 | -6.731 0 | C246 | -0.511 6 | C470 | -0.229 1 | ||||||
| B465 | 7.411 0 | C560 | -0.853 8 | C485 | -1.425 9 | ||||||
| B516 | 33.917 8 | C250 | 0.504 5 | C500 | 0.330 6 | ||||||
| B460 | 15.365 3 | C260 | -4.067 7 | C242 | -1.334 4 | ||||||
| B270 | 48.274 5 | C262 | 2.209 | ||||||||
| TC1A02 | C475 | -15.280 2 | C447 | -1.908 5 | C575 | 2.100 0 | |||||
| C500 | 7.374 5 | GM1843 | E111 | -3.135 8 | C580 | -1.821 0 | |||||
| C591 | -29.501 6 | E117 | -0.238 5 | C245 | -3.147 3 | ||||||
| C470 | 41.588 5 | E157 | -2.066 7 | C565 | 2.648 1 | ||||||
| C485 | -13.043 2 | E119 | 2.327 5 | C246 | -0.807 5 | ||||||
| C500 | -51.089 | E121 | 3.294 1 | C560 | -2.600 1 | ||||||
| C242 | 38.863 2 | E132 | -2.161 2 | C250 | -2.266 5 | ||||||
| C575 | 17.545 3 | C260 | -3.740 9 | ||||||||
| C580 | 20.984 1 | C262 | -1.317 6 | ||||||||
| C245 | -39.861 6 | C447 | -1.123 5 | ||||||||
| C565 | -28.856 2 | GM1843 | E111 | -2.869 5 | |||||||
| C246 | -37.283 | E117 | -0.629 2 | ||||||||
| C560 | 40.926 2 | E157 | 4.316 6 | ||||||||
| C250 | -62.524 5 | E119 | 0.728 9 | ||||||||
| C260 | -32.857 4 | E121 | 2.357 2 | ||||||||
| C262 | -18.781 8 | E132 | -0.283 7 | ||||||||
| C447 | -21.705 4 | AAE (positive/negative) | |||||||||
| AAE (positive/negative) | AAE (positive/negative) | PM54 | 1.074 9 | -2.205 2 | |||||||
| PM54 | 34.132 4 | -35.064 6 | GM529 | 0.885 6 | -6.695 | GM529 | 0.566 2 | -5.755 9 | |||
| pPGPseq2E6 | 22.565 6 | -18.911 5 | TC1A02 | 2.241 2 | -2.713 5 | TC1A02 | 1.619 4 | -1.958 5 | |||
| TC1A02 | 27.880 3 | -31.889 4 | GM1843 | 2.810 8 | -1.900 6 | GM1843 | 2.467 6 | -1.260 8 | |||
| 1 | 盖钧镒, 章元明, 王建康 (2003). 植物数量性状遗传体系. 北京: 科学出版社. |
| 2 | 高运来, 姚丙晨, 刘春燕, 李文福, 蒋洪蔚, 李灿东, 张闻博, 胡国华, 陈庆山 (2009). 黑龙江省主栽大豆品种遗传多样性的SSR分析. 植物学报 44, 556-561. |
| 3 | 黄莉, 任小平, 张晓杰, 陈玉宁, 姜慧芳 (2012). ICRISAT花生微核心种质农艺性状和黄曲霉抗性关联分析. 作物学报 38, 935-946. |
| 4 | 赖明芳, 曾彦, 漆燕, 夏友霖, 崔富华 (2007). 花生主要经济性状遗传特点分析. 中国油料作物学报 29, 148-151. |
| 5 | 李兰周, 刘风珍, 万勇善, 张昆, 赵文祥 (2013). 花生荚果和籽仁相关性状的主基因+多基因混合遗传模型分析. 华北农学报 28(5), 116-123. |
| 6 | 李孟军, 肖寒, 卢金东, 王兴军 (2008). 花生微卫星标记的研究进展. 植物学通报 25, 373-380. |
| 7 | 刘华 (2011). 栽培花生产量和品质相关性状遗传分析与QTL定位研究. 硕士论文. 郑州: 河南农业大学. |
| 8 | 吕维娜 (2014). 花生栽培种SSR遗传连锁图谱构建及重要产量性状QTL定位分析. 硕士论文. 郑州: 郑州大学. |
| 9 | 孙大容 (1998). 花生育种学. 北京: 中国农业出版社. pp. 55. |
| 10 | 谭贤杰, 吴子恺, 程伟东, 王天宇, 黎裕 (2011). 关联分析及其在植物遗传学研究中的应用. 植物学报 46, 108-118. |
| 11 | 万建民 (2006). 作物分子设计育种. 作物学报 32, 455-462. |
| 12 | 万书波 (2003). 中国花生栽培学. 上海: 上海科学技术出版社. pp. 78-79. |
| 13 | 王建康, 李慧慧, 张学才, 尹长斌, 黎裕, 马有志, 李新海, 邱丽娟, 万建民 (2011). 中国作物分子设计育种. 作物学报 37, 191-201. |
| 14 | 文自翔, 赵团结, 郑永战, 刘顺湖, 王春娥, 王芳, 盖钧镒 (2008). 中国栽培和野生大豆农艺品质性状与SSR标记的关联分析I. 群体结构及关联标记. 作物学报 34, 1169-1178. |
| 15 | 吴学君 (2010). 我国花生及花生产品出口结构与竞争力分析——基于1997-2007年进出口贸易数据. 中国油料作物学报 32, 309-314. |
| 16 | 严玫 (2012). 花生种质资源重要农艺和品质性状的全基因组关联分析. 硕士论文. 郑州: 郑州大学. |
| 17 | 杨小红, 严建兵, 郑艳萍, 余建明, 李建生 (2007). 植物数量性状关联分析研究进展. 作物学报 33, 523-530. |
| 18 | 禹山林 (2008). 中国花生品种及其系谱. 上海: 上海科学技术出版社. pp. 232-701. |
| 19 | 张新友 (2010). 栽培花生产量、品质和抗病性的遗传分析与QTL定位研究. 博士论文. 杭州: 浙江大学. |
| 20 | 张新友, 韩锁义, 徐静, 严玫, 刘华, 汤丰收, 董文召, 黄冰艳 (2012). 花生主要品质性状的QTLs定位分析. 中国油料作物学报 34, 311-315. |
| 21 | Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME (1993). Optimizing parental selection for genetic linkage maps.Genome 36, 181-186. |
| 22 | Belamkar V, Selvaraj MG, Ayers JL, Payton PR, Puppala N, Burow MD (2011). A first insight into population structure and linkage disequilibrium in the US peanut minicore collection.Genetica 139, 411-429. |
| 23 | Bradbury PJ, Zhang ZW, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007). TASSEL: software for association mapping of complex traits in diverse samples.Bioinformatics 23, 2633-2635. |
| 24 | Breseghello F, Sorrells ME (2006). Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars.Genetics 172, 1165-1177. |
| 25 | Evanno G, Regnaut S, Goudet J (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study.Mol Ecol 14, 2611-2620. |
| 26 | Falush D, Stephens M, Pritehard JK (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies.Genetics 164, 1567-1587. |
| 27 | FAO (2011, 2012). Statistical database. Rome: Food and Agriculture Organization of the United Nations. |
| 28 | Gupta PK, Rustgi S, Kulwal PL (2005). Linkage disequilibrium and association studies in higher plants: pres- ent status and future prospects.Plant Mol Biol 57, 461-485. |
| 29 | Hong YB, Liang XQ, Chen XP, Liu HY, Zhou GY, Li SX, Wen SJ (2008). Construction of genetic linkage map based on SSR markers in peanut (Arachis hypogaea L.). Agric Sci China 7, 915-921. |
| 30 | Jin L, Lu Y, Xiao P, Sun M, Corke H, Bao JS (2010). Genetic diversity and population structure of a diverse set of rice germplasm for association mapping.Theor Appl Genet 121, 475-487. |
| 31 | Kraakman ATW, Niks RE, Van den Berg PMMM, Stam P, Van Eeuwijk FA (2004). Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars.Genetics 168, 435-446. |
| 32 | Krapoviekas A (1969). The origin, variability and spread of the groundnut (Arachis hypogaea L.). In: Ucko PJ, Dimbleby GW, eds. The Domestication and Exploitation of Plants and Animals. London: Duckworth. pp. 427-441. |
| 33 | Li XH, Acharya A, Farmer AD, Crow JA, Bharti AK, Kramer RS, Wei YL, Han YH, Gou JQ, May GD, Monteros MJ, Brummer EC (2012). Prevalence of single nucleotide polymorphism among 27 diverse alfalfa genotypes as assessed by transcriptome sequencing.BMC Genomics 13, 568. |
| 34 | Liu KJ, Muse SV (2005). PowerMarker: an integrated analysis environment for genetic marker analysis.Bioinformatics 21, 2128-2129. |
| 35 | Mackay I, Powell W (2007). Methods for linkage disequilib- rium mapping in crops.Trends Plant Sci 12, 57-63. |
| 36 | Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J, Kreitman M, Maloof JN, Noyes T, Oefner PJ, Stahl EA, Weigel D (2002). The extent of linkage disequilibrium inArabidopsis thaliana. Nat Genet 30, 190-193. |
| 37 | Nordborg M, Hu TT, Ishino Y, Jhaveri J, Toomajian C, Zheng HG, Bakker E, Calabrese P, Gladstone J, Goyal R, Jakobsson M, Kim SK, Morozov Y, Padhuka- sahasram B, Plagnol V, Rosenberg NA, Shah C, Wall JD, Wang J, Zhao KY, Kalbfleisch T, Schulz V, Kreitman M, Bergelson J (2005). The pattern of polymorphism in Arabidopsis thaliana.PLoS Biol 3, e196. |
| 38 | Porebski S, Bailey LG, Baum BR (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components.Plant Mol Biol Rep 15, 8-15. |
| 39 | Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data.Genetics 155, 945-959. |
| 40 | Ren XP, Jiang HF, Yan ZY, Chen YN, Zhou XP, Huang L, Lei Y, Huang JQ, Yan LY, Qi Y, Wei WH, Liao BS (2014). Genetic diversity and population structure of the major peanut (Arachis hypogaea L.) cultivars grown in China by SSR markers.PLoS One 9, e88091. |
| 41 | Templeton AR (1995). A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping or DNA sequencing. V. Analysis of case/control sampling designs: alzheimer's disease and the apoprotein E locus.Genetics 140, 403-409. |
| 42 | Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ESIV (2001). Dwarf8 polymorphisms associate with variation in flowering time.Nat Genet 28, 286-289. |
| 43 | Wang ML, Zhu CS, Barkley NA, Chen ZB, Erpelding JE, Murray SC, Tuinstra MR, Tesso T, Pederson GA, Yu JM (2009). Genetic diversity and population structure analysis of accessions in the US historic sweet sorghum collection.Theor Appl Genet 120, 13-23. |
| 44 | Yang XH, Yan JB, Shah T, Warburton ML, Li Q, Gao YF, Chai YC, Fu ZY, Zhou Y, Xu ST, Bai GH, Meng YJ, Zheng YP, Li JS (2010). Genetic analysis and characterization of a new maize association mapping panel for quantitative trait loci dissection.Theor Appl Genet 121, 417-431. |
| 45 | Yu JM, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doe- bley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006). A unified mixed- model method for association mapping that accounts for multiple levels of relatedness.Nat Genet 38, 203-208. |
| 46 | Zhao KY, Aranzana MJ, Kim S, Lister L, Shindo C, Tang C, Toomajian C, Zheng HG, Dean C, Nordborg M (2007). An Arabidopsis example of association mapping in structured samples.PLoS Genet 3, e4. |
| No related articles found! |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
 首页
首页